Lipoid Proteinosis and Heritable Disorders of Connective Tissue: Introduction
Inherited connective tissue disorders are clinically and genetically diverse conditions affecting the skin, joints, and a variety of extracutaneous tissues, including the cardiovascular (CV) system. The nature and severity of the skin phenotype is dependent on the type of mutation as well as the role of the affected protein on dermal structure and function (see Chapter 63). The diagnosis of many inherited connective tissue disorders is clinical, although CLIA-approved genetic testing is increasingly available. A listing of research laboratories that may offer testing can be found at http://www.genetests.org.
Ehlers–Danlos Syndrome
|
A variety of genetically and clinically heterogeneous inherited conditions characterized by varying degrees of skin fragility and hyperextensibility, joint hypermobility, and easy bruising, have been collected under the rubric of the Ehlers–Danlos syndromes (EDS). A new classification system was proposed in 19971 to consolidate and simplify existing subgroups (Table 137-1). Despite these revisions, many patients defy clear-cut classification due to overlapping features. The spectrum of EDS disease severity ranges from nearly imperceptible findings to severe, debilitating disease. As such, it is difficult to estimate EDS prevalence, but if milder forms are included it may be as high as 1:5,000 individuals. The kyphoscoliosis, arthrochalasia, and dermatosparaxis types are considerably less common than the classical, hypermobility, and vascular types. Confusion over the diagnosis of the hypermobility type and its overlap with joint hypermobility syndrome interferes with prevalence estimates. If inclusive definitions are used, it may be the most common subtype. Generalized joint hypermobility is the consistent and unifying finding in all forms of EDS.3 EDS types other than the common classic type and the life-threatening vascular type are discussed in the online edition of the book.
Villefranche Type/(OMIM) | Clinical Features | Inheritance | Protein/(Gene Defect) |
|---|---|---|---|
Classical/(130000 and 130010) | Hyperextensible skin; easy bruising; wide, atrophic scars; hypermobile joints | AD | Collagen type V/(COL5A1, COL5A2) |
Hypermobility/(130020) | Smooth, velvety skin; joint hypermobility | AD/AR | Unclear for most; collagen type III; tenascin XB/(COL3A1; TNXB)a |
Vascular/(130050) | Thin, translucent skin with easy bruising; arterial and visceral rupture; typical facies | AD | Collagen type III/(COL3A1) |
Kyphoscoliosis/(225400 and 229200) | Atrophic scars, easy bruising; neonatal hypotonia; scoliosis; ocular rupture; marfanoid habitus | AR | Lysyl hydroxylase/(PLOD1) |
Arthrochalasia/(130060) | Hyperextensible and fragile skin; severe joint hypermobility; congenital hip dislocation | AD | Collagen type I/(COL1A1; COL1A2) |
Dermatosparaxis/(225410) | Severely fragile, sagging, redundant skin; hernias and premature rupture of fetal membranes | AR | Procollagen I N-peptidase/(ADAMTS2) |
Other typesa | Wrinkled, loose facial skin; curly fine hair; scanty eyebrows and eyelashes | AR | Due to mutations in galactosyltransferase |
Progeroid variant2/(130070) |
Classical EDS occurs in 1 in 10,000–20,000 newborns.4
Approximately half of classical EDS cases are caused by autosomal dominant (AD) defects in the α1 or α2 chain of type V collagen (COL5A1 and COL5A2). Type V collagen is a minor fibrillar collagen that regulates collagen fibril diameter. These mutations may result in dominant negative or haploinsufficient states and approximately one-third of cases are due to haploinsufficiency of the COL5A1 gene.5 Recently, mutations in the signal peptide region of COL5A1, which disrupt type V collagen secretion, have also been identified in patients with classical EDS.6 An additional locus may exist, as several families do not exhibit linkage to COL5A1 or COL5A2.7,8
Patients with features of classic EDS exhibiting defective type I collagen due to AD COL1A1 mutations have also been described.9 These patients have nonglycine substitutions in the type I collagen triple helical domain and, during adulthood, some have developed vascular complications, including spontaneous arterial rupture, similar to the complications of the vascular EDS subtype.10 An autosomal recessive (AR) “cardiac valvular subtype of EDS” exhibits clinical findings of classic EDS as well as the onset of severe cardiac valve problems later in life and results from total loss of the proa2(I) chains of type I collagen due to homozygous or compound heterozygous mutations in COL1A2.11 An AR form of EDS resembling a mild version of classic EDS can result from homozygous tenascin-X (TNX) deficiency. Heterozygotes (especially females) for these mutations may exhibit hypermobility EDS.12
Electron microscopy of the skin in EDS reveals thickened collagen fibrils, highlighting the role of type V collagen in regulating their size. It is proposed that the amino terminus of α1(V) carries a negative charge, conferred by abundant tyrosine residues, and appears to limit fibril growth. Less than 5% of fibrils may exhibit “collagen cauliflowers,” which are rare composite fibrils.13–15
Patients with classic EDS typically exhibit some degree of skin and joint hyperextensibility, tissue fragility, and bruising. In the past, patients exhibiting very mild features were termed “mitis.” Typical patients have hyperextensible skin (Fig. 137-1) that recoils easily to its normal position after stretching, in contrast to the skin in cutis laxa (CL). Skin hyperextensibility should be assessed by pulling until resistance is met at a site not subjected to mechanical forces or scarring, such as the volar surface of the forearm. The normal upper limit of extensibility at the volar forearm is 1–1.5 cm. Skin overlying extensor joints tends to be redundant and should not be used to assess extensibility.
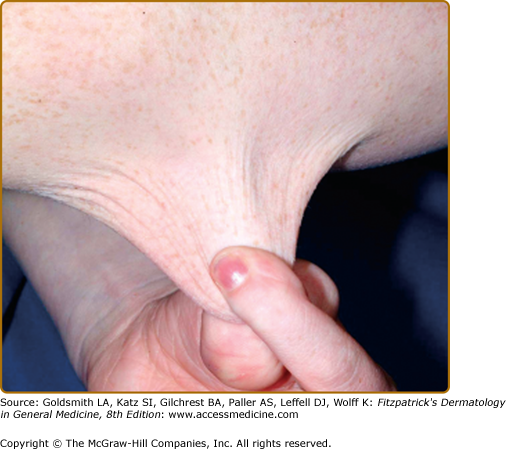
The skin is velvety, thin, and bruises easily. Fetuses with classical EDS may exhibit growth retardation, hernias, and joint dislocations. Bruising manifests early in childhood and is often persistent in areas prone to trauma in young children, especially the shins (Fig. 137-2) Tissue fragility can be striking, with seemingly innocuous trauma resulting in disproportionately large skin tears that are relatively painless without excessive bleeding. Wounds often gape.
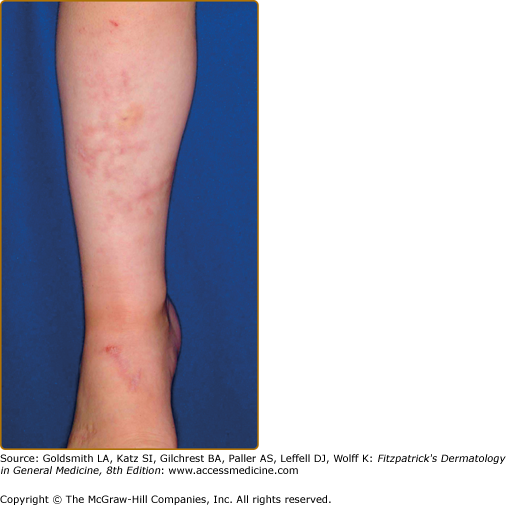
Even with surgical repair, wounds in EDS often exhibit slow healing and frequent infection. Postoperative wound dehiscence is common if wounds are not adequately secured, often necessitating repeated repairs or secondary intention healing (Fig. 137-3). Even with surgical repair, dehiscence upon suture removal may occur. Healed wounds result in atrophic, often widened cigarette paper-like (or “fish mouth”) scars, especially on pressure points (knees, elbows, forehead, chin) (Figs. 137-2 and 137-3). Hematoma formation, especially at pressure points, often results in persistent hyperpigmentation. Calcification and fibrosis of hematomas produce subcutaneous, nodular molluscoid pseudotumors, most frequently around the elbow and knee. These are often associated with scars. Spheroids are small subcutaneous spherical hard nodules, usually on the forearms and shins that may become calcified and thus detectable radiographically. Additionally, subcutaneous fat herniation on the medial or lateral aspects of the heels or wrists with pressure (piezogenic pedal papules) may be seen. Acrocyanosis and chilblains have also been described in affected individuals.
Figure 137-3
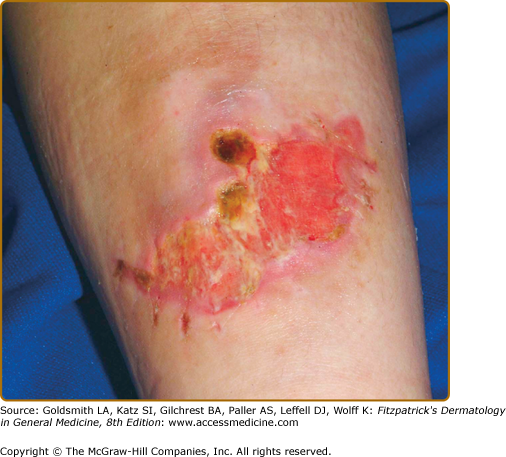
Classical Ehlers–Danlos syndrome. After suturing for a laceration, the wound dehisced with secondary infection and marked widening. Note the evidence of former sutures at the lower border, now 3 weeks after the injury and treatment with antibiotics. Scars tend to stretch further in the 6 months after closure.
Joint hypermobility is frequent (Fig. 137-4), and often greatest at the fingers and/or wrists. In suspected patients, including older children and adolescents, it is assessed using the Beighton scale Table 137-2).1,3 Sprains, dislocations or subluxations, scoliosis, and pes planus are common complications and patients frequently describe chronic joint and limb pain, although skeletal radiographs may be normal.16 Patients are typically “double-jointed” and often able to perform various “tricks” with their joints as a result. Such chronic and excessive joint hypermobility frequently leads to early-onset osteoarthritis. Osteoporosis is more common when compared to age-matched controls. Muscle hypotonia and delayed gross motor development are also described. EDS may carry a diagnosis of chronic fatigue syndrome or fibromyalgia due to persistent fatigue and chronic pain.
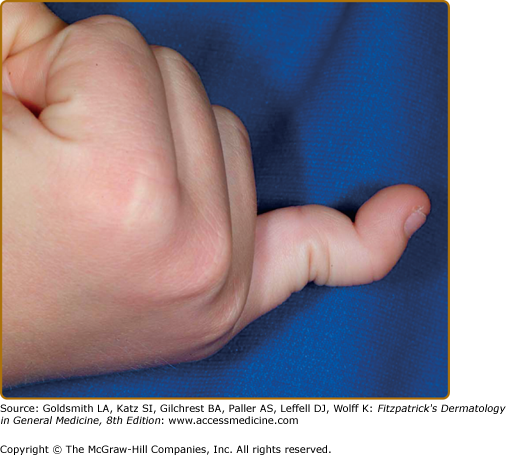
|
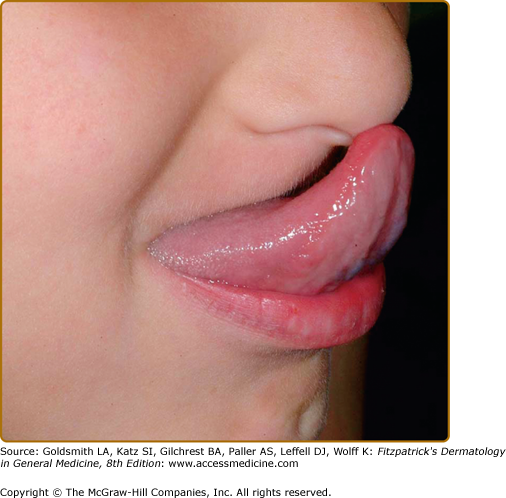
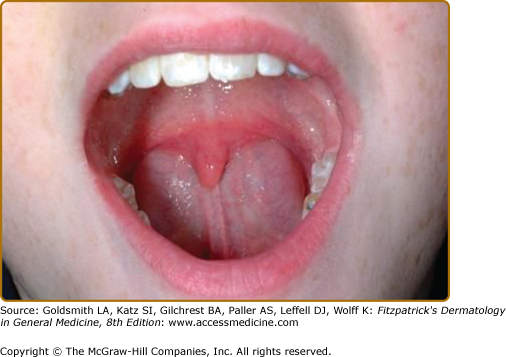
Most patients with classic or hypermobile EDS are able to extend the tongue to touch the tip of the nose (Gorlin’s sign) (Fig. 137-5), although this sign can be displayed by ∼10% of individuals without inherited disorders of connective tissue. Lack of lingual and labial frenulae has recently been noted to be a minor feature of EDS, and is particularly common in patients with the vascular type17 Hypermobility and absence of the frenulae allow some EDS patients to “swallow” their own tongue (eFig. 137-5.1). Hiatal and postoperative hernias as well as anal prolapse have been noted as manifestations of the tissue hyperextensibility and fragility.18 Sexual dysfunction, including dyspareunia is reported, and functional bowel disorders occur in up to half of patients.19
Approximately 40%–50% of affected individuals (in addition to ∼20% of infants born to affected mothers) are born prematurely (32–37 weeks), either due to premature rupture of fetal membranes or chorioamnionitis.20,21 Monitoring during pregnancy and the postpartum period is recommended, because of the risk of premature labor during the third trimester, the increased risk for skin tears, postpartum hemorrhage, and uterine/bladder prolapse.19 The fragility of the already abnormal connective tissue may be worsened by the effect of pregnancy-related hormones that soften connective tissue, such as relaxin.
Patients with classical EDS occasionally exhibit structural heart malformations. Mitral valve prolapse (MVP), and less often tricuspid valve prolapse, may occur and recent reports suggest that aortic root dilatation may occur more frequently than past studies indicated.22 Arterial rupture, intracranial aneurysms, and arteriovenous fistulae have been described in classic EDS, typically in patients exhibiting severe clinical phenotypes, but are much less common than in Marfan syndrome. Baseline echocardiography with measurement of aortic diameter is recommended,19,22 with CT or MRI exams if needed.16 Longitudinal data on progression of aortic dilatation in classic EDS do not exist. The current recommendation is yearly echocardiogram if cardiac abnormalities (aortic dilatation, MVP) are present. If no abnormalities are noted, subsequent studies are only recommended as dictated by symptoms. β-blocker therapy has been successful anecdotally.
 Hypermobility EDS is thought to be relatively common (1:10,000–15,000 individuals),4 although estimates may be inaccurate due to underdiagnosis. The relationship between the so-called “benign-familial articular hypermobility syndrome” or “joint hypermobility syndrome” and hypermobility EDS is currently a matter of controversy due to the lack of availability of confirmatory laboratory testing and the wide range of phenotypic severity of hypermobility EDS. If all individuals with joint hypermobility syndrome are included, hypermobility EDS would be the most common inherited connective tissue disorder.
Hypermobility EDS is thought to be relatively common (1:10,000–15,000 individuals),4 although estimates may be inaccurate due to underdiagnosis. The relationship between the so-called “benign-familial articular hypermobility syndrome” or “joint hypermobility syndrome” and hypermobility EDS is currently a matter of controversy due to the lack of availability of confirmatory laboratory testing and the wide range of phenotypic severity of hypermobility EDS. If all individuals with joint hypermobility syndrome are included, hypermobility EDS would be the most common inherited connective tissue disorder.
 The molecular basis for hypermobility EDS is largely unclear. One family was found to have haploinsufficiency of COL3A1.23 Approximately 5% of patients with the hypermobility form show diminished levels of tenascin X (TNX) from heterozygous mutations in the tenascin-XB (TNX-B) gene, which encodes TNX24 (see Chapter 63). The phenotype in these patients is intermediate between classical EDS and hypermobility EDS; approximately 40% of patients exhibit abnormal skin findings similar to classical EDS but without atrophic scarring.25 The inheritance of hypermobility EDS is typically autosomal dominant (AD), although the rare cases with tenascin-XB mutations are autosomal recessive (AR).1 The association of hypermobility EDS with congenital adrenal hyperplasia has been reported, owing to recombination within the TNX gene and CYP21 deletion.13 A quadricuspid aortic valve and midline defects (such as bifid uvula) have also been described in association.26
The molecular basis for hypermobility EDS is largely unclear. One family was found to have haploinsufficiency of COL3A1.23 Approximately 5% of patients with the hypermobility form show diminished levels of tenascin X (TNX) from heterozygous mutations in the tenascin-XB (TNX-B) gene, which encodes TNX24 (see Chapter 63). The phenotype in these patients is intermediate between classical EDS and hypermobility EDS; approximately 40% of patients exhibit abnormal skin findings similar to classical EDS but without atrophic scarring.25 The inheritance of hypermobility EDS is typically autosomal dominant (AD), although the rare cases with tenascin-XB mutations are autosomal recessive (AR).1 The association of hypermobility EDS with congenital adrenal hyperplasia has been reported, owing to recombination within the TNX gene and CYP21 deletion.13 A quadricuspid aortic valve and midline defects (such as bifid uvula) have also been described in association.26
 The diagnosis of hypermobility EDS is clinical and based on appropriate exam and family history.27 Phenotypic findings are quite variable and many patients who seek medical care are female. Patients with hypermobility EDS show soft, velvety skin with occasional mild hyperextensibility but no fragility. Patients often note easy bruising and piezogenic papules may be seen. In occasional patients mild prolongation of bleeding can mimic von Willebrand disease. Scarring is typically normal, thus if healed wounds are atrophic, the classic type should be diagnosed instead. Molluscoid pseudotumors are not seen.
The diagnosis of hypermobility EDS is clinical and based on appropriate exam and family history.27 Phenotypic findings are quite variable and many patients who seek medical care are female. Patients with hypermobility EDS show soft, velvety skin with occasional mild hyperextensibility but no fragility. Patients often note easy bruising and piezogenic papules may be seen. In occasional patients mild prolongation of bleeding can mimic von Willebrand disease. Scarring is typically normal, thus if healed wounds are atrophic, the classic type should be diagnosed instead. Molluscoid pseudotumors are not seen.
 Joint hypermobility is generalized and dislocations of the shoulder, patella, and temporomandibular joints are particularly common. The hips and digits may also be involved and scoliosis and pes planus are also seen. Musculoskeletal pain often has an early onset, is chronic, and often debilitating. Fatigue and sleep disturbances are typical and hypermobility EDS patients are often diagnosed with a variety of psychiatric conditions or fibromyalgia. Headaches are common. Temporomandibular joint disorder is more frequent28 and a pain syndrome (regional) has been associated with hypermobility EDS, which may result from chronic stretch injury to the nerves passing over hypermobile joints as well as fragility of the neural connective tissue.29 Depression is common in these patients, often related to their chronic pain.30 Early onset of osteoarthritis may occur and osteoporosis is more common than in age-matched controls. Functional bowel disorders may occur in up to 50% of patients. Pelvic prolapse and dyspareunia have also been noted.31
Joint hypermobility is generalized and dislocations of the shoulder, patella, and temporomandibular joints are particularly common. The hips and digits may also be involved and scoliosis and pes planus are also seen. Musculoskeletal pain often has an early onset, is chronic, and often debilitating. Fatigue and sleep disturbances are typical and hypermobility EDS patients are often diagnosed with a variety of psychiatric conditions or fibromyalgia. Headaches are common. Temporomandibular joint disorder is more frequent28 and a pain syndrome (regional) has been associated with hypermobility EDS, which may result from chronic stretch injury to the nerves passing over hypermobile joints as well as fragility of the neural connective tissue.29 Depression is common in these patients, often related to their chronic pain.30 Early onset of osteoarthritis may occur and osteoporosis is more common than in age-matched controls. Functional bowel disorders may occur in up to 50% of patients. Pelvic prolapse and dyspareunia have also been noted.31
 One-third to one-half of hypermobility EDS patients will note some type of atypical chest pain, heart palpitations, and/or orthostatic intolerance. Aortic root dilatation can occur in 25%–33% of patients; however, it is usually mild and in the absence of significant dilatation its long-term risk is unclear. Increased elastic fiber degeneration relative to unaffected controls has been demonstrated on electron microscopy32 and dilatation and/or rupture of the ascending aorta has been described in these patients.33
One-third to one-half of hypermobility EDS patients will note some type of atypical chest pain, heart palpitations, and/or orthostatic intolerance. Aortic root dilatation can occur in 25%–33% of patients; however, it is usually mild and in the absence of significant dilatation its long-term risk is unclear. Increased elastic fiber degeneration relative to unaffected controls has been demonstrated on electron microscopy32 and dilatation and/or rupture of the ascending aorta has been described in these patients.33
 A proposed course of clinical progression of hypermobility EDS has recently been described. Infants first show the “hypermobility” phase, which by the second decade shifts to a “pain” phase, during which joint hypermobility decreases (although still hypermobile by the Beighton scale), but worsening pain progressively impacts quality of life and function. The third phase, “stiffness,” is characterized by progressive limitation of joint mobility and a dramatic impact on the quality of life. Depression and anxiety are common during this phase. Additional findings in these patients are dysphonia, headache, and dolichocolon.34
A proposed course of clinical progression of hypermobility EDS has recently been described. Infants first show the “hypermobility” phase, which by the second decade shifts to a “pain” phase, during which joint hypermobility decreases (although still hypermobile by the Beighton scale), but worsening pain progressively impacts quality of life and function. The third phase, “stiffness,” is characterized by progressive limitation of joint mobility and a dramatic impact on the quality of life. Depression and anxiety are common during this phase. Additional findings in these patients are dysphonia, headache, and dolichocolon.34
This is the most clinically significant EDS subtype due to the risk of arterial or major organ rupture. Inherited in an AD fashion, the incidence is approximately 1 in 100,000–200,000 individuals.4
Vascular EDS is associated with dominant negative mutations in the type III collagen gene (COL3A1), resulting in reduced amounts of type III collagen in the dermis, vessels, and viscera, as well as decreased production and secretion of type III collagen by cultured fibroblasts.35 More than 320 different mutations have been reported (exon-skipping or missense) and typically lead to disruption of the triple-helical structure of type III collagen. As type III collagen is a homotrimer, mutant proα1(III) chains affect most fibrils, interfering with secretion, and leading to intracellular accumulation. Type III collagen plays an important role in the integrity of both blood vessel walls and the upper dermis. Analysis of type III procollagen and collagen chains and direct genetic testing of COL3A1 have been utilized to test for vascular EDS.
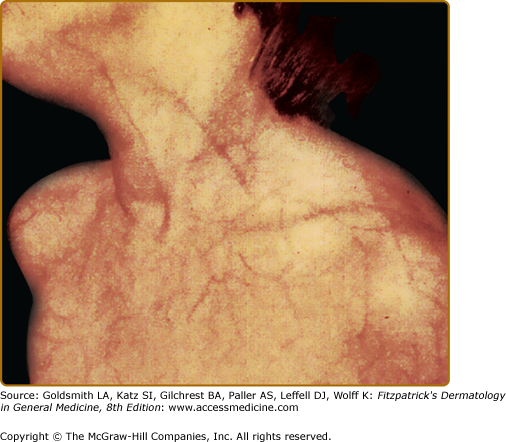
The classic quadrad of vascular type of EDS includes a characteristic facial appearance (which may be subtle), thin, translucent skin with a prominent venous pattern (Fig. 137-6), extensive bruising or hematomas, and vascular or visceral rupture (or both).18 Joint hypermobility is usually minimal and limited to the digits. The facial features include a thin nose and upper lip, small earlobes, and sunken, pigmented periocular regions. Subcutaneous fat is decreased, especially of the face and limbs. Widened, thin (papyraceous) scars may be noted on bony prominences. Hematomas, which may be frequent and/or quite large, may develop after minimal to no trauma, such as sphygmomanometer inflation.
Spontaneous rupture of arteries, particularly midsized arteries, may occur during childhood, although its peak age of incidence is the third or fourth decade of life. Mesenchymal abdominal, splenic, and renal arteries are often involved, as is the descending aorta. Aortic rupture/dissection is typically not preceded by detectable aortic dilatation,22 occurs distal to the midaortic arch, and exhibits distal extension. Arterial or intestinal rupture often presents as acute abdominal or flank pain; arterial rupture is the most common cause of death. Stroke is also reported in vascular EDS. Pregnancies may be complicated by pre- and postpartum arterial bleeding, and by intrapartum uterine rupture. Pregnancies carry a 12%–25% fatality rate20,36 Vaginal and perineal tears from delivery heal poorly and caesarean wounds often dehisce.4
 This extremely rare subtype of EDS has been described in approximately 60 patients worldwide.4 Inheritance is AR and clinical findings are congenital.
This extremely rare subtype of EDS has been described in approximately 60 patients worldwide.4 Inheritance is AR and clinical findings are congenital.
 Mutations in the lysyl hydroxylase (PLOD1 or procollagenlysine, 2-oxoglutarate 5-dioxygenase) gene cause the kyphoscoliotic form of EDS. Lysyl hydroxylase functions to hydroxylate lysine residues present in collagen. This defect results in decreased hydroxylysine content in the dermis of kyphoscoliotic EDS patients.37 Hydroxylysine residues are a prominent site of cross-link formation between adjacent collagen molecules and their deficiency leads to abnormal cross-linking. Lysyl hydroxylase deficiency prominently affects only certain collagen and tissue types, typically both type I and type III collagen.38 An intragenic duplication of exons 9–15 appears to be a relatively common cause of kyphoscoliotic EDS, affecting 19% of families39 A rare form of unknown cause exhibits the typical clinical findings, but normal lysyl hydroxylase activity and skin hydroxylysine content.40
Mutations in the lysyl hydroxylase (PLOD1 or procollagenlysine, 2-oxoglutarate 5-dioxygenase) gene cause the kyphoscoliotic form of EDS. Lysyl hydroxylase functions to hydroxylate lysine residues present in collagen. This defect results in decreased hydroxylysine content in the dermis of kyphoscoliotic EDS patients.37 Hydroxylysine residues are a prominent site of cross-link formation between adjacent collagen molecules and their deficiency leads to abnormal cross-linking. Lysyl hydroxylase deficiency prominently affects only certain collagen and tissue types, typically both type I and type III collagen.38 An intragenic duplication of exons 9–15 appears to be a relatively common cause of kyphoscoliotic EDS, affecting 19% of families39 A rare form of unknown cause exhibits the typical clinical findings, but normal lysyl hydroxylase activity and skin hydroxylysine content.40
 Patients with kyphoscoliosis type EDS exhibit congenital progressive kyphoscoliosis and generalized joint laxity with severe muscle hypotonia, leading to gross motor delay. Loss of the ability to ambulate is common by the second or third decade of life. The skin may be fragile and heals with atrophic scarring. The associated hyperextensibility is less prominent than that of classical EDS. Easy bruisability and arterial rupture have been noted. Occasionally, patients show a Marfanoid habitus, osteopenia, and scleral fragility with rupture of the ocular globe.
Patients with kyphoscoliosis type EDS exhibit congenital progressive kyphoscoliosis and generalized joint laxity with severe muscle hypotonia, leading to gross motor delay. Loss of the ability to ambulate is common by the second or third decade of life. The skin may be fragile and heals with atrophic scarring. The associated hyperextensibility is less prominent than that of classical EDS. Easy bruisability and arterial rupture have been noted. Occasionally, patients show a Marfanoid habitus, osteopenia, and scleral fragility with rupture of the ocular globe.
 While it is possible to measure lysyl hydroxylase activity using cultured fibroblasts, the more widely used diagnostic screening test is detection by high-performance liquid chromatography of a ratio of urinary deoxypyridinoline to pyridinoline cross-links, that is, at least one order of magnitude greater than age-matched controls.41,42
While it is possible to measure lysyl hydroxylase activity using cultured fibroblasts, the more widely used diagnostic screening test is detection by high-performance liquid chromatography of a ratio of urinary deoxypyridinoline to pyridinoline cross-links, that is, at least one order of magnitude greater than age-matched controls.41,42
 The arthrochalasia-type of EDS is extremely rare with only ∼30 cases reported.4 Inheritance is AD. The type B subtype appears to be more common.43
The arthrochalasia-type of EDS is extremely rare with only ∼30 cases reported.4 Inheritance is AD. The type B subtype appears to be more common.43
 Both the arthrochalasia and dermatosparaxis (see below) forms involve impaired removal of the amino terminal N-propeptides from type I procollagen. In the arthrochalasia-type, mutations in the type I collagen genes (COL1A1 or COL1A2) lead to removal of exon 6 during mRNA processing (exon-skipping). The result is deficient processing of the amino terminal propeptide ends of the proα1(I) (type A) or proα2(I) (type B) chains of collagen I and thus impaired cross-linking of type I collagen.44–46
Both the arthrochalasia and dermatosparaxis (see below) forms involve impaired removal of the amino terminal N-propeptides from type I procollagen. In the arthrochalasia-type, mutations in the type I collagen genes (COL1A1 or COL1A2) lead to removal of exon 6 during mRNA processing (exon-skipping). The result is deficient processing of the amino terminal propeptide ends of the proα1(I) (type A) or proα2(I) (type B) chains of collagen I and thus impaired cross-linking of type I collagen.44–46
 Affected individuals show profound generalized joint hypermobility with recurrent subluxations of both large and small joints, which are difficult to repair surgically.47 Congenital bilateral hip dislocation is considered a hallmark of this subtype, although it may also be seen in the classical and vascular forms. The skin may be hyperextensible, easy to bruise, and fragile with widened scars, although milder than in classical EDS. Short stature, muscle hypotonia, kyphoscoliosis, and mild osteopenia have been described.48 Affected infants are often born in a breech position.43
Affected individuals show profound generalized joint hypermobility with recurrent subluxations of both large and small joints, which are difficult to repair surgically.47 Congenital bilateral hip dislocation is considered a hallmark of this subtype, although it may also be seen in the classical and vascular forms. The skin may be hyperextensible, easy to bruise, and fragile with widened scars, although milder than in classical EDS. Short stature, muscle hypotonia, kyphoscoliosis, and mild osteopenia have been described.48 Affected infants are often born in a breech position.43
 The normal triple helix of mature collagen is composed of two α1 and one α2 molecules. Thus, mutations in COL1A1 in arthrochalasia type A leads to a more severe effect on collagen fibrils, which exhibit a strikingly ragged outline (due to 75% of fibrils having one or two abnormal α1 collagen components). In contrast, type B with mutations in COL1A2 lead to collagen fibrils with a wide variation in diameter but smoother outlines.43 The detection of pNα1(I) or pNα2(I) chains using electrophoresis of extracted patient collagen may be used to confirm the diagnosis. Detection of exon 6 skipping in cDNA of COL1A1 or COL1A2 may also be utilized, with mutation analysis in positive cases.46
The normal triple helix of mature collagen is composed of two α1 and one α2 molecules. Thus, mutations in COL1A1 in arthrochalasia type A leads to a more severe effect on collagen fibrils, which exhibit a strikingly ragged outline (due to 75% of fibrils having one or two abnormal α1 collagen components). In contrast, type B with mutations in COL1A2 lead to collagen fibrils with a wide variation in diameter but smoother outlines.43 The detection of pNα1(I) or pNα2(I) chains using electrophoresis of extracted patient collagen may be used to confirm the diagnosis. Detection of exon 6 skipping in cDNA of COL1A1 or COL1A2 may also be utilized, with mutation analysis in positive cases.46
 Dermatosparaxis EDS is the rarest EDS subtype, with only ∼10 reported cases in humans, although well described in cattle.49 Inheritance is AR.
Dermatosparaxis EDS is the rarest EDS subtype, with only ∼10 reported cases in humans, although well described in cattle.49 Inheritance is AR.
 The dermatosparaxis form results from homozygous or compound heterozygous mutations in the procollagen I N-proteinase gene (ADAMTS-2).49 Absence of procollagen I N-proteinase results in failure to remove the amino terminal propeptide of type I collagen.49–51 Collagen fibrils are dramatically distorted in dermatosparaxis EDS and are often said to resemble hieroglyphics.43 Electrophoresis of dermal collagen (extracted in presence of protease inhibitors) or from fibroblasts demonstrates type I collagen procollagen α1(I) and α2(I) chains.1
The dermatosparaxis form results from homozygous or compound heterozygous mutations in the procollagen I N-proteinase gene (ADAMTS-2).49 Absence of procollagen I N-proteinase results in failure to remove the amino terminal propeptide of type I collagen.49–51 Collagen fibrils are dramatically distorted in dermatosparaxis EDS and are often said to resemble hieroglyphics.43 Electrophoresis of dermal collagen (extracted in presence of protease inhibitors) or from fibroblasts demonstrates type I collagen procollagen α1(I) and α2(I) chains.1
 The dermatosparaxis type of EDS form is characterized by severe skin fragility with sagging, redundant skin that is not extensible.52 Wound healing is not impaired and scars are not atrophic. The skin may be soft and doughy in texture, with easy bruisability. Patients exhibit large fontanels, puffy eyelids, short fingers, and blue sclera. Large umbilical and inguinal hernias, and premature rupture of the fetal membranes may be seen.53 Additional findings described in dermatosparaxis-type EDS include micrognathia, dental malocclusion, and hyperkeratotic gingival hyperplasia. Abnormal molars and severe dental enamel defects of the deciduous teeth are reported. Abnormalities of the permanent teeth include microdontia, discoloration, root dysplasia, obliteration of dental pulp, and agenesis.
The dermatosparaxis type of EDS form is characterized by severe skin fragility with sagging, redundant skin that is not extensible.52 Wound healing is not impaired and scars are not atrophic. The skin may be soft and doughy in texture, with easy bruisability. Patients exhibit large fontanels, puffy eyelids, short fingers, and blue sclera. Large umbilical and inguinal hernias, and premature rupture of the fetal membranes may be seen.53 Additional findings described in dermatosparaxis-type EDS include micrognathia, dental malocclusion, and hyperkeratotic gingival hyperplasia. Abnormal molars and severe dental enamel defects of the deciduous teeth are reported. Abnormalities of the permanent teeth include microdontia, discoloration, root dysplasia, obliteration of dental pulp, and agenesis.
 Other types of EDS have been described in single families or are associated with features that may be serendipitous.54 Mutations in the amino-terminal region of type I collagen interfere with N-terminal propeptide removal (without disrupting N-proteinase cleavage site), and result in an EDS/osteogenesis imperfecta (OI) overlap phenotype.55 Patients with progeroid EDS (OMIM #130070) exhibit a phenotype of loose, wrinkled facial skin, fine curly hair, sparse eyebrows and eyelashes, downslanting palpebral fissures, developmental delay, skeletal abnormalities, as well as cutaneous findings typical of EDS. The progeroid variant has been attributed to mutations in galactosyltransferase I.56 Periventricular heterotopia (PH), a neuronal migration defect that may manifest as seizures, has been reported in patients meeting clinical criteria for EDS. Mutations in filamin A have been detected in a subset of these patients with PH and EDS, which appears to be an X-linked dominant disorder. These patients have an increased risk of arterial rupture from aortic root dilatation, as well as joint hypermobility, recurrent joint dislocations, increased bruising, and abnormal scarring.57
Other types of EDS have been described in single families or are associated with features that may be serendipitous.54 Mutations in the amino-terminal region of type I collagen interfere with N-terminal propeptide removal (without disrupting N-proteinase cleavage site), and result in an EDS/osteogenesis imperfecta (OI) overlap phenotype.55 Patients with progeroid EDS (OMIM #130070) exhibit a phenotype of loose, wrinkled facial skin, fine curly hair, sparse eyebrows and eyelashes, downslanting palpebral fissures, developmental delay, skeletal abnormalities, as well as cutaneous findings typical of EDS. The progeroid variant has been attributed to mutations in galactosyltransferase I.56 Periventricular heterotopia (PH), a neuronal migration defect that may manifest as seizures, has been reported in patients meeting clinical criteria for EDS. Mutations in filamin A have been detected in a subset of these patients with PH and EDS, which appears to be an X-linked dominant disorder. These patients have an increased risk of arterial rupture from aortic root dilatation, as well as joint hypermobility, recurrent joint dislocations, increased bruising, and abnormal scarring.57
 A recently described series of patients exhibited characteristic craniofacial features, progressive skin and joint laxity, multiple congenital contractures (fingers, wrists, hips), as well as cardiac, gastrointestinal (GI), respiratory, and ocular abnormalities. Despite some similarities clinically to kyphoscoliotic EDS, lysyl hydroxylase was normal and the underlying defect remains unclear. Skin findings included bruisability, fragility, healing with atrophic scars, recurrent subcutaneous infections with fistula formation, and recurrent hematomas.58
A recently described series of patients exhibited characteristic craniofacial features, progressive skin and joint laxity, multiple congenital contractures (fingers, wrists, hips), as well as cardiac, gastrointestinal (GI), respiratory, and ocular abnormalities. Despite some similarities clinically to kyphoscoliotic EDS, lysyl hydroxylase was normal and the underlying defect remains unclear. Skin findings included bruisability, fragility, healing with atrophic scars, recurrent subcutaneous infections with fistula formation, and recurrent hematomas.58
 The spondylocheirodysplastic form of EDS (SCD-EDS) exhibits clinical features similar to type VI EDS, including translucent, easily bruisable hyperelastic skin, small joint hypermobility with a predisposition to eventual contractures, protuberant eyes with bluish sclerae, tapering fingers, thenar atrophy, and finely wrinkled palmar skin. Skeletal abnormalities in these patients include platyspondyly, moderate short stature, osteopenia, and widened metaphyses. Mutations in a membrane-bound zinc transporter, SLC39A13 (OMIM #608735), have been identified. The disrupted zinc (Zn2+) transport may lead to increased endoplasmic reticulum zinc concentrations. Competition of the increased Zn2+ with Fe2+, a necessary cofactor for hydroxylation of lysyl and prolyl residues on collagen, results in the underhydroxylated lysyl and prolyl residues seen in these patients, and leads to impaired collagen cross-linking and decreased triple helical stability.59
The spondylocheirodysplastic form of EDS (SCD-EDS) exhibits clinical features similar to type VI EDS, including translucent, easily bruisable hyperelastic skin, small joint hypermobility with a predisposition to eventual contractures, protuberant eyes with bluish sclerae, tapering fingers, thenar atrophy, and finely wrinkled palmar skin. Skeletal abnormalities in these patients include platyspondyly, moderate short stature, osteopenia, and widened metaphyses. Mutations in a membrane-bound zinc transporter, SLC39A13 (OMIM #608735), have been identified. The disrupted zinc (Zn2+) transport may lead to increased endoplasmic reticulum zinc concentrations. Competition of the increased Zn2+ with Fe2+, a necessary cofactor for hydroxylation of lysyl and prolyl residues on collagen, results in the underhydroxylated lysyl and prolyl residues seen in these patients, and leads to impaired collagen cross-linking and decreased triple helical stability.59
Routine histopathologic examination of the skin from EDS patients is typically normal. Electron microscopy demonstrates abnormalities in the appearance of collagen fibrils as noted above. Despite the bruising and increased bleeding, tests of platelet function and coagulation are usually normal, further highlighting the underlying defects in skin and blood vessel structural integrity. As noted above for specific subtypes, while genetic diagnosis is possible, especially as a research tool, a variety of biochemical tests are available as well.
EDS must be distinguished clinically and histologically from CL. In CL, the hyperelastic skin does not return to its normal position after stretching. The kyphoscoliotic and hypermobility forms of EDS must be distinguished from Marfan syndrome, which additionally is characterized by ectopia lentis and characteristic skeletal abnormalities. Molluscoid pseudotumors of the lower legs may mimic subcutaneous granuloma annulare. The easy bruisability and poor wound healing of EDS patients has led to concerns of child abuse.
Vascular EDS exhibits significant clinical similarities to Loeys–Dietz syndrome type 2, an aortic aneurysm syndrome caused by mutations in transforming growth factor (TGF)-β receptor 1 and 2 (TGFBR1 and TGFBR2) genes. These patients also exhibit skin findings including velvety translucent skin, easy bruising, and widened, atrophic scars.60 Prior to detection of arterial anomalies Loeys–Dietz syndrome patients may be misdiagnosed as classic or hypermobility EDS.
 Patients with X-linked occipital horn syndrome exhibit lax skin and joints, vascular tortuosity, bladder diverticulae, and hernias in addition to the eponymous wedge-shaped calcifications on the occipital bone. However, the skin does not show easy bruising or fragility. X-linked occipital horn syndrome is caused by mutations in the ATP7A copper transporter and is allelic to Menkes disease. Serum copper and ceruloplasmin are low.
Patients with X-linked occipital horn syndrome exhibit lax skin and joints, vascular tortuosity, bladder diverticulae, and hernias in addition to the eponymous wedge-shaped calcifications on the occipital bone. However, the skin does not show easy bruising or fragility. X-linked occipital horn syndrome is caused by mutations in the ATP7A copper transporter and is allelic to Menkes disease. Serum copper and ceruloplasmin are low.
 Ullrich disease, also known as scleroatonic muscular dystrophy, is a congenital disorder resulting from AD or AR defects in type VI collagen. Affected patients often exhibit striking distal joint hyperextensibility, in addition to proximal contractures and general muscle weakness.25
Ullrich disease, also known as scleroatonic muscular dystrophy, is a congenital disorder resulting from AD or AR defects in type VI collagen. Affected patients often exhibit striking distal joint hyperextensibility, in addition to proximal contractures and general muscle weakness.25
 Patients with Stickler syndrome can exhibit early-onset osteoarthritis, scoliosis, and joint abnormalities. However, they also demonstrate characteristic ocular (vitreoretinal) abnormalities, sensorineural hearing loss, and cleft palate. Mutations in three collagen genes (COL2A1, COL11A1, and COL11A2) result in Stickler syndrome.61,62
Patients with Stickler syndrome can exhibit early-onset osteoarthritis, scoliosis, and joint abnormalities. However, they also demonstrate characteristic ocular (vitreoretinal) abnormalities, sensorineural hearing loss, and cleft palate. Mutations in three collagen genes (COL2A1, COL11A1, and COL11A2) result in Stickler syndrome.61,62
 Williams syndrome (Williams–Beuren syndrome; OMIM #194050) is caused by a contiguous gene deletion of the Williams–Beuren syndrome critical region on chromosome 7q11.23, which includes the gene for elastin. During childhood these patients may exhibit lax joints and decreased muscle tone, although with age joint stiffness/contractures may develop. Specific cardiovascular (CV) disease is present, including an elastin arteriopathy, supravalvular aortic stenosis (in cases with mutation but not deletion of the elastin gene), and peripheral pulmonic stenosis. Patients exhibit a characteristic facies, mental retardation (typically mild), short stature, low birthweight and slow weight gain along with feeding difficulties during infancy. Patients with Williams syndrome often exhibit a very characteristic personality, being quite sociable with a strong affinity for music. Endocrine abnormalities such as hypercalcemia, hypercalciuria, precocious puberty, and hypothyroidism also occur.63
Williams syndrome (Williams–Beuren syndrome; OMIM #194050) is caused by a contiguous gene deletion of the Williams–Beuren syndrome critical region on chromosome 7q11.23, which includes the gene for elastin. During childhood these patients may exhibit lax joints and decreased muscle tone, although with age joint stiffness/contractures may develop. Specific cardiovascular (CV) disease is present, including an elastin arteriopathy, supravalvular aortic stenosis (in cases with mutation but not deletion of the elastin gene), and peripheral pulmonic stenosis. Patients exhibit a characteristic facies, mental retardation (typically mild), short stature, low birthweight and slow weight gain along with feeding difficulties during infancy. Patients with Williams syndrome often exhibit a very characteristic personality, being quite sociable with a strong affinity for music. Endocrine abnormalities such as hypercalcemia, hypercalciuria, precocious puberty, and hypothyroidism also occur.63
 Patients with Aarskog–Scott syndrome (faciodigitogenital syndrome, shawl scrotum syndrome, faciogenital dysplasia; OMIM #305400) may also exhibit hyperextensibility, especially of the finger joints, as well as hypermobility of the cervical spine and scoliosis. However, other typical features of the syndrome, including facial dysmorphic features, shawl scrotum, and X-linked recessive (XLR) inheritance allow its differentiation.64,65
Patients with Aarskog–Scott syndrome (faciodigitogenital syndrome, shawl scrotum syndrome, faciogenital dysplasia; OMIM #305400) may also exhibit hyperextensibility, especially of the finger joints, as well as hypermobility of the cervical spine and scoliosis. However, other typical features of the syndrome, including facial dysmorphic features, shawl scrotum, and X-linked recessive (XLR) inheritance allow its differentiation.64,65
 Patients with the macrocephaly, alopecia, cutis laxa (CL), and scoliosis (MACS) syndrome exhibit skin hyperextensibility rather than laxity, and thus the “cutis laxa” in MACS may be a misnomer.66 The elastic tissue abnormalities in MACS are limited to the papillary dermis. This AR disorder results from mutations in Ras and Rab interactor 2 protein (RIN2). Ubiquitously expressed, RIN2 interacts with Rab5 to regulate endocytic trafficking, which appears to alter function of the Golgi apparatus. The Golgi apparatus in these patients accumulates vacuoles, and evaluation of dermis shows few dermal microfibrils and deficiency of fibulin-5 (also deficient in CL, see Section “Clinical Findings” under “Pseudoxanthoma Elasticum”).67
Patients with the macrocephaly, alopecia, cutis laxa (CL), and scoliosis (MACS) syndrome exhibit skin hyperextensibility rather than laxity, and thus the “cutis laxa” in MACS may be a misnomer.66 The elastic tissue abnormalities in MACS are limited to the papillary dermis. This AR disorder results from mutations in Ras and Rab interactor 2 protein (RIN2). Ubiquitously expressed, RIN2 interacts with Rab5 to regulate endocytic trafficking, which appears to alter function of the Golgi apparatus. The Golgi apparatus in these patients accumulates vacuoles, and evaluation of dermis shows few dermal microfibrils and deficiency of fibulin-5 (also deficient in CL, see Section “Clinical Findings” under “Pseudoxanthoma Elasticum”).67
A multidisciplinary preventative strategy is the most productive approach to the management of EDS patients. Early identification of affected patients with appropriate intervention (pain management, physical therapy, surgery) and education is important.68–70 Nonweight-bearing exercise (swimming) can promote muscle development. Exercise regimens are typically designed to strengthen muscles to stabilize joints and relieve stress. Physical therapy focused on shoulder girdle strengthening, has decreased the frequency of shoulder dislocations in patients with a history of chronic or recurrent dislocation. Some patients use orthopedic devices such as orthotics and braces with benefit.
Avoiding and preventing injuries is of great importance. Assessment of the home environment with modifications to make homes “EDS safe,” such as avoiding hard, sharp edges on furniture and arrangement to prevent or minimize falls, can be helpful. Patients with significant skin fragility or bruising may require protective padding or bandaging and should avoid contact sports and heavy exercise.
When cutaneous wounds do occur, they should be sutured using both subcuticular and cuticular sutures, which are tightly spaced and left in place for a prolonged duration (at least twice as long as standard). Adhesive tapes, bolsters, or pressure bandages are necessary to aid healing, diminish scarring, and lower the risk of hematoma and pseudotumor formation. Close monitoring for postoperative infection is critical and preventative antibiotic therapy is often employed. Pseudotumors of the elbows or knees are typically more easily surgically removed than those on the heels.
Ongoing rheumatologic and orthopedic care may be required to prevent progressive joint disease in certain patients. Low-impact sports are preferable to contact sports and weight training in patients with hypermobility EDS. Ascorbate therapy (∼2 gm/day in adults with proportional decreases in pediatric patients) may improve bruising (but not other findings) in classic EDS. Anti-inflammatory drugs may improve the musculoskeletal pain associated with EDS, but those interfering with platelet function (especially aspirin) should be avoided in patients with significant bruising. Patients with valvular heart disease should be followed by cardiologists, and a baseline echocardiogram with measurement of aortic diameter is recommended before 10 years of age.25
Joint dislocation (with the exception of congenital dislocation of the hip) in EDS typically spontaneously resolves or can be remedied with closed reduction. Patients with hypermobility EDS may benefit from a variety of physical therapy interventions, such as myofascial release therapies and low-resistance exercises for muscle toning. Exercise progression should involve increasing the number of repetitions, frequency, or duration of exercise, rather than increasing resistance and should progress slowly. Pain from writing utensils can be lessened by altering the grip to rest the shaft of writing utensils on the thenar web and holding the tip between the index and ring fingers. Transvaginal pelvic physical therapy has been successfully utilized for abdominal and back pain as well as radicular pain of the lower extremities and dyspareunia. Braces are commonly used to stabilize joints. Intervention is typically multidisciplinary and should involve rheumatology, orthopedic surgery, physical and occupational therapy, and pain management specialists as appropriate. Pain in hypermobility EDS is often undertreated. While reparative or corrective surgeries are often delayed as long as possible, the perioperative complication rate in patients with hypermobility EDS is not increased. Gastrointestinal (GI) symptoms may be quite troublesome and require aggressive therapy with proton pump inhibitors, H2-blockers, and other agents. Calcium and vitamin D supplementation is typically encouraged.31
Patients with vascular EDS should refrain from contact sports or isometric exercise and avoid medications, which impair platelet function. The traditional recommendation has been that invasive vascular procedures should be avoided, unless absolutely necessary (such as with arterial rupture), due to the extremely high risk of vascular rupture. When performed, tissues must be handled with extreme caution due to pronounced intraoperative tissue fragility. A recent series suggested early intervention by skilled surgeons and provided suggestions for management.71 Studies examining the use of β-blockers in vascular EDS are in progress.
Fatigue is a common finding in EDS of most types, present in over three-fourth of patients. It appears most common in hypermobility EDS, but is seen in other forms as well. Contributing factors include disruptions of sleep, impaired self-efficacy, and pain.72 A variety of neuromuscular findings, including reduction in vibration sense and mild-to-moderate muscle weakness, were commonly found in a small group of multiple types of EDS. These symptomatic findings correlated with myopathic features on needle electromyography (EMG) (a mixed neurogenic–myopathic pattern in 60%) as well as muscle ultrasound showing increased echo-intensity and atrophy. The subset of patients with hypermobility EDS resulting from TNX-B haploinsufficiency seem to have fewer neuromuscular issues, although TNX levels correlate inversely with neuromuscular symptoms.73
Pregnancy in the EDS patient should be considered high-risk. The effects of hormonally mediated connective tissue softening on the abnormal connective tissue of EDS have not been studied, but may account for the increased skin fragility, as well as other EDS sequelae, that occur during pregnancy and the immediate postpartum period. Prenatal diagnosis is possible by genetic and biochemical analyses. Unless children are severely affected by their disorder, they generally adjust well to the skin findings and joint hypermobility. The Ehlers–Danlos Foundation is a national support group (www.ednf.org).70
Marfan Syndrome
|
Marfan syndrome is an AD disorder primarily affecting the skeletal, ocular, and CV systems.74 About 25% of cases occur sporadically, particularly in patients born of older fathers. Parental germline mosaicism has been described.75 The overall prevalence of Marfan syndrome is estimated at approximately 1:5,000–10,000 persons with no racial, gender, or geographic predilection.76 The prognosis for patients with Marfan syndrome has improved over the past three decades due to better medical and surgical treatments.77
While standard histopathology of affected skin appears normal, with electron microscopy dermal collagen in Marfan syndrome exhibits abnormal thickness, array, and shape, including fibrils of variable thickness, disarray, twisted fibrils, and fibrils with a flower-like appearance and ragged margins, similar to those found in EDS. The content ratio of collagen I to collagen III is also decreased.32 Recently, defects in the architecture of the collagen microfibrils of large vessels, rather than collagen content or cross-linking, have been described.78 The resultant architecture is similar to that seen in abdominal aortic aneurysms and is proposed to explain the weakened aneurysmal vessels in Marfan syndrome.
Marfan syndrome results from heterozygous mutations79 in the profibrillin 1 gene on chromosome 15q21.1.80 The spectrum of clinical phenotypes resulting from FBN1 defects extends beyond classic Marfan syndrome and these disparate conditions have been termed fibrillopathies/fibrillinopathies81 These range from the severe neonatal Marfan phenotype to isolated aortic root dilatation or marfanoid skeletal features without typical CV pathology or ectopia lentis. Fibrillin is a 350-kDa glycoprotein that is a major component of extracellular matrix (ECM) microfibrils, which are structural components of the zonular fibers of the suspensory ligament of the lens and associated with elastic fibers in the aorta and skin. Virtually every family’s mutation is different and over 500 mutations have been reported occurring in any of the 65 exons of the gene. No hot spots have been identified, except in cases of neonatal Marfan syndrome. Interestingly, mutations creating premature termination codons appear to lead to milder disease, while those in exons 24–31 are associated with neonatal Marfan syndrome and more severe disease.76,82 Marfan syndrome type II is caused by mutations at the TGFBR2 gene locus.
Fibrillin interacts with latent TGF-β binding protein, sequestering it, and controlling TGF-β availability. Deficiency of fibrillin increases TGF-β availability and fibrillin-1 mutation leads to constitutive activation of latent TGF-β with signaling through both “classic” and noncanonical TGF-β pathways. In vascular tissues, TGF-β is believed to be a critical mediator of both the structure and the function of the vascular ECM, via both matrix deposition and degradation. Increased TGF-β signaling enhances ECM degradation, leading to aneurysm development and progression. Stimulation of either TGF-β pathway can also lead to phosphorylation of Smad, which triggers a profibrotic signaling response. Angiotensin II signals through two receptor types, type 1 (AT1) and type 2 (AT2), which exhibit opposite signaling effects. Angiotensin II signaling through AT1 also increases expression of TGF-β ligands and receptors and stimulates vessel wall fibrosis. In addition to the action of activated TGF-β, macroaggregates of fibrillin monomers form the basic scaffold on which mature elastin fibers are assembled; as a result, abnormal fibrillin creates a disordered microfibril matrix that may further lead to disordered and weak elastic fiber formation and disruption of the microfibril network that connects elastic lamellae to neighboring interstitial cells.83
Research into the impact of these pathways and their potential therapeutic implications for Marfan syndrome is ongoing. Losartan, a blocker of angiotensin II type 1 receptor, has already shown promise in slowing the aortic root dilatation in individuals with Marfan syndrome. Losartan not only blocks TGF-β but appears to decrease phosphorylation of Smad2 independent of its TGF-β effects. Thus, AT1 blockade may interrupt several pathways, which affect vascular remodeling and disease progression.84
Marfan syndrome is a generalized connective tissue disorder exhibiting abnormalities of three primary organ systems: ocular (typically lens dislocation); skeletal (excessive extremity length, loose joints, anterior chest deformities, and kyphoscoliosis); and most importantly, CV (classically aortic aneurysm and mitral valve redundancy).81 Many of the typical physical features of Marfan syndrome are age-dependent, making diagnosis in childhood more difficult. No single clinical sign is pathognomonic.85
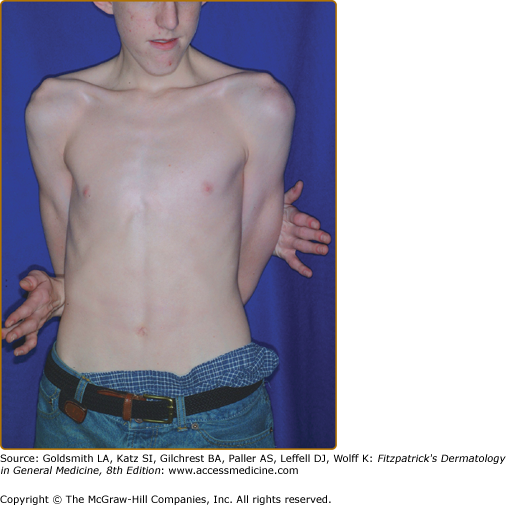
The “Marfanoid habitus” is characteristically dolichostenomelic (tall and thin), with a lower body segment (pubic symphysis to floor) that is longer than the upper segment (height minus lower segment) (Fig. 137-7). Characteristically, the arm span exceeds the person’s height by several centimeters. The distal bones are excessively long (arachnodactyly). Skeletal features of Marfan syndrome are characterized by bone overgrowth and joint laxity. Kyphoscoliosis may be severe and increases with the adolescent growth spurt.86 Thoracic cage abnormalities, such as pectus excavatum (sternal depression) or carinatum (sternal projection), both result from excessive rib overgrowth and are common. The combination of kyphoscoliosis and pectus excavatum rarely compromises cardiopulmonary function. Joint laxity from capsular, ligamentous, and tendinous involvement may cause flat feet, knee or elbow hyperextensibility (genu recurvatum), and occasional joint dislocation. Patellar dislocation is not uncommon; dislocation of the hip, often detected during the newborn period, may be the first sign of Marfan syndrome. Screening tests for joint hypermobility are the thumb (or Steinberg) sign, in which the thumb extends well beyond the ulnar border of the hand when overlapped by the fingers, and the wrist (or Walker–Murdoch) sign, in which the thumb overlaps the fifth finger as they grasp the opposite wrist.86 The underlying joint hyperextensibility and long extremities of Marfan patients often enable them to reach around their back and touch their umbilicus from the opposite side.
Most patients with Marfan syndrome exhibit myopia due to flattening of the corneas and an abnormally long anterior–posterior orbital axis.87 An estimated 50%–70% of patients have ectopia lentis, typically with upward lens displacement. Subluxation and complete dislocation of the lens often lead to secondary ocular abnormalities, including ametropia, myopia, acute glaucoma, and increased risk of retinal detachment.75 As mild displacements may be missed with standard ocular exams, referral to ophthalmology for a dilated slit lamp exam is necessary if a diagnosis of Marfan syndrome is suspected.
CV abnormalities are responsible for the majority of the morbidity and mortality in Marfan patients. CV abnormalities are detected in ∼40% of patients with Marfan syndrome by cardiac examination and almost 100% of patients by autopsy examination. Medial necrosis of the aorta is the most common defect and diffuse dilatation of the proximal segment of the ascending aorta with aortic regurgitation often occurs. Such dilatation is progressive, and may even be detected in utero. The progression of dilatation is not always continuous and this unpredictability mandates frequent monitoring.88 Death in Marfan patients usually occurs in adulthood as a result of CV sequelae, most commonly secondary to dilatation of the aortic root,89,90 leading to aortic dissection or rupture and pericardial tamponade. MVP results from dilation of the mitral valve annulus, with stretching of the chordae and MV leaflet redundancy. It occurs in ∼25% of affected children and adolescents, and in 86% with associated pectus excavatum.91 MVP increases with age, and eventually occurs in ∼75% of patients.92 MVP may lead to abnormal findings on electrocardiogram, mitral valvular regurgitation, and even cardiac arrhythmias leading to sudden death.74
Lack of subcutaneous fat and the presence of striae, most prominent on the upper chest, arms, thighs, and abdomen, are the most common cutaneous manifestations of Marfan syndrome. These cutaneous features are found in up to two-third of patients. Elastosis perforans serpiginosa (see Chapter 69) is more common in individuals with Marfan syndrome, and inguinal or incisional hernias may occur. Skin findings are considered minor diagnostic features in the revised diagnostic criteria.93
Dural ectasia (stretching of the dural sac in the lumbosacral region) often develops in patients with Marfan syndrome. Emphysema has been described, and lung bullae increase the risk of pneumothorax, particularly involving the upper lobes. Oral findings may include a high-arched palate and crowding of anterior teeth.
Infants with the neonatal form of Marfan syndrome have the body disproportion of Marfan syndrome in addition to lax skin, emphysema, ocular abnormalities, joint contractures, kyphoscoliosis, adducted thumbs, crumpled ears, micrognathia, muscle hypoplasia, and deficient subcutaneous fat over joints.94,95 Severe cardiac valve insufficiency and aortic dilatation result in death during the first 2 years of life.
A recent large retrospective study highlighted clinical features of children in a cohort of more than 1,000 patients with FBN1 mutations. The median age of diagnosis was 6.5 years, and only 30% had a positive family history. Patients with neonatal Marfan syndrome represented 14% of the cohort. Of the affected children, 19% were noted to be severely affected, 32% to have classic Marfan, and 35% of the patients to have probable Marfan syndrome. In the pediatric population, 71% already exhibited dilatation of the ascending aorta. Ectopia lentis was present in approximately half and typical marfanoid skeletal findings in 28%. Aortic arch dilatation was more common in neonatal cases; however, aortic complications were rare in childhood. In uncertain cases the identification of FBN1 mutations often aided diagnosis. While neonatal Marfan syndrome represents a severe de novo early-onset form with associated high mortality, patients diagnosed after adolescence typically resembled adult patients in phenotype. Milder cases were more often had a positive family history, likely because milder cases are more commonly survived to reproductive age. Importantly, pediatric patients often did not meet international criteria for the diagnosis of Marfan syndrome at the time of first examination. As the features of the syndrome increase with age, follow-up examination is critical in suspected pediatric cases to identify affected patients.76
The diagnosis of Marfan syndrome is based on a constellation of clinical findings. There is no diagnostic laboratory test or histologic abnormality. Increased urinary hydroxyproline and desmosine are not consistent findings. Ophthalmologic examination by an experienced ophthalmologist with interventions as necessary should be performed as early as possible. Orthopedic evaluation as needed and echocardiographic monitoring of aortic root size and valvular function is paramount with cardiology involvement depending on the findings.
Marfan syndrome is rarely confused with disorders other than homocystinuria (see online edition of this book). However, a variety of conditions exhibit partial phenotypic overlap with Marfan syndrome, including some also caused by fibrillin-1 mutations. These are summarized in Box 137-1.
Disorder | Clinical Similarities to Marfan | Clinical Differences from Marfan | Defect |
|---|---|---|---|
Homocystinuria | Dolichostenomelia Orthopedic abnormalities Ectopia lentis (downward displacement) | Elevated fasting homocysteine levels AR | Cystathionine β synthetase deficiency causes abnormal methionine metabolism and increased levels of urinary homocysteine |
MASS (Mitral valve prolapse; Aortic root diameter; Stretch marks; Skeletal features) syndrome | Mitral valve prolapse Myopia Skin—striae Skeletal findings AD | Borderline aortic enlargement is not progressive | Heterozygous fibrillin 1 mutations |
Mitral valve prolapse (MVP) syndrome | Mitral valve prolapse Skeletal findings AD | Lack of other features | Fibrillin 1 |
Familial ectopia lentis | Ectopia lentis Skeletal changes | Questionable risk of eventual aortic enlargement: Current recommendations are for periodic imaging | Heterozygous fibrillin 1 mutations |
Shprintzen–Goldberg syndrome | Dolichostenomelia Arachnodactyly Scoliosis Pectus Highly arched palate Occasional aortic root enlargement | Craniosynostosis Developmental delay Hypertelorism Proptosis Rib anomalies Chiari malformation Equinovarus deformity | Fibrillin mutations not present in most cases |
Loeys–Dietz syndrome (LDS) | Generalized arterial tortuosity with aneurysms and dissection occurring throughout Aneurysms more labile than Marfan syndrome Early dissections and ruptures even in childhood AD | No ectopia lentis Dolichostenomelia less frequent/obvious Hypertelorism Cleft palate with broad/bifid uvula Learning disabilities Chiari I malformation Hydrocephalus Blue sclerae Craniosynostosis Talipes Exotropia Soft, velvety, translucent, easily bruised skin | Mutations in TGFBR1 and TGFBR2 have been detected |
Familial thoracic aortic aneurysms and aortic dissection (FTAAD) syndrome | Vascular disease AD | No other typical clinical features | TGFBR2 mutations and other loci |
Ehlers–Danlos syndrome (see above) | Kyphoscoliotic form EDS may exhibit increased risk for rupture of medium-sized arteries Vascular EDS has joint laxity (often only small joints), translucent skin, characteristic facies, organ rupture, and tendency for aneurysm or dissection of medium to large muscular arteries AD | Vascular pathology not limited to the aortic root, although this may be involved | |
Weil–Marchesani syndrome | Ectopia lentis | Orthopedic features differ Short stature Brachydactyly Spherophakia Stiff joints | |
Congenital contractural arachnodactyly (CCA; Beals syndrome) | Congenital contractures (elbows, knees, hips, and fingers)—improve with time Ocular (keratoconus) and/or cardiovascular (aortic root dilatation, mitral valve prolapse, and septal defects) findings rare Arachnodactyly, progressive severe kyphoscoliosis High, arched palate Muscular hypoplasia/weakness | Characteristic folded upper helix—“crumpled” appearance of ear—improves with time | Defects in second fibrillin gene (FBN2– typically in the “neonatal region” of exons 23–34)96 |
Management of Marfan syndrome has focused on prevention of the disabling and life-threatening potential complications. Early and regular ophthalmologic examinations are required to detect correctable amblyopia and retinal detachment. Ectopia lentis and even complete subluxation may be tolerated for decades. Lens extraction may be required to treat diplopia, glaucoma, cataracts, or retinal detachment. Lasix surgery is contraindicated.
Repair of pectus excavatum is appropriate if cardiopulmonary compromise develops, but is delayed until skeletal maturation is nearly complete to prevent recurrence and should utilize internal stabilization.99 Scoliosis may be lessened in adolescent girls by estrogen therapy, but this may produce an overall decrease in height. Bracing, physical therapy, and vertebral fusion may all be required to prevent severe scoliosis.
A family history of early aortic dissection mandates aggressive monitoring. At a minimum, patients with Marfan syndrome should undergo yearly monitoring. Aortic complications have not been reported in patients with an aortic diameter normal for age or less than 40 mm in diameter (in adults)74 Long-term propranolol therapy has been administered to prevent aortic dilatation by decreasing myocardial contractility,100 but may not affect survival.101 In a small number of children with Marfan syndrome who were recalcitrant to β-blockers, angiotensin II type 1 receptor blockers (losartan and irbesartan) led to a significant decrease in aortic root diameter and dilation of the sinotubular junction. Larger randomized trials examining the effect of these agents are underway.102 Aneurysmal and valvular heart defects may require prosthetic replacement, but this should be delayed for as long as possible to avoid recurrent prosthesis replacement, particularly in growing children. Replacement of the aortic root has led to increased life expectancy and is indicated once the maximal measurement is greater than 5 cm in adults and older children, the rate of size increase is ∼1 cm/year, or progressive aortic regurgitation develops. Patients with known aortic dilatation should avoid caffeine, stressful circumstances, and vigorous exercise. Patients with pulmonary involvement should avoid situations with rapid changes in air pressure, such as scuba diving or flying and, of course, should not smoke.103
Doxycycline inhibits matrix metalloproteinase and has been shown to improve aortic wall architecture and delay aortic dissection in mouse models. Future studies may focus on synergy between doxycycline and losartan, as matrix metalloproteinases can activate TGF-β.77
Children should be excused from participation in physical education in order to avoid potentially harmful exertion, contact sports, and isometric exercises, which might lead to aortic rupture or congenital heart failure. Unfortunately, this can add to the isolation of a child who may already be concerned about an unusual body image or is socially ostracized because of looking “different” or being excessively tall. Importantly, not all patients with Marfan syndrome exhibit striking “classic” phenotypes. When the diagnosis in a patient with an atypical presentation is suspected, one must make certain that appropriate studies are performed, so that potentially lethal internal manifestations are not neglected. The Web site for the National Marfan Foundation is www.marfan.org.
Homocystinuria
|
 The incidence of homocystinuria is estimated at 1:200,000–350,000 live births worldwide and it is considered the second most common treatable aminoaciduria after phenylketonuria. In Ireland, a founder effect has led to an incidence of ∼1:65,000.104,105
The incidence of homocystinuria is estimated at 1:200,000–350,000 live births worldwide and it is considered the second most common treatable aminoaciduria after phenylketonuria. In Ireland, a founder effect has led to an incidence of ∼1:65,000.104,105
 Recently, a report from Denmark identified a lower actual incidence of homocystinuria (diagnosis based on elevated methionine levels at the time of newborn screening) than would be expected based on population frequency. Incidence rates based on newborn screening are suspected to underestimate incidence, as B6-responsive patients may not have elevated methionine levels at the time of newborn screening. More recently molecular screening studies have allowed estimation of the incidence of homozygosity for the B6-responsive c.833T>C (p.1278T) allele that may account for ∼21% of cystathionine β-synthase (CBS) inactivating alleles worldwide. Testing in 500 Danish newborns suggested a 1.4% heterozygosity rate for this mutation, which would predict a homozygosity incidence of 1:20,400—far more frequent than the 1:344,000 incidence rate currently accepted. This discrepancy suggests that some patients with homozygous CBS deficiency may be missed and raises the possibility that there are mildly affected patients who are not diagnosed.106 Further studies in different populations are necessary to clarify this discrepancy.
Recently, a report from Denmark identified a lower actual incidence of homocystinuria (diagnosis based on elevated methionine levels at the time of newborn screening) than would be expected based on population frequency. Incidence rates based on newborn screening are suspected to underestimate incidence, as B6-responsive patients may not have elevated methionine levels at the time of newborn screening. More recently molecular screening studies have allowed estimation of the incidence of homozygosity for the B6-responsive c.833T>C (p.1278T) allele that may account for ∼21% of cystathionine β-synthase (CBS) inactivating alleles worldwide. Testing in 500 Danish newborns suggested a 1.4% heterozygosity rate for this mutation, which would predict a homozygosity incidence of 1:20,400—far more frequent than the 1:344,000 incidence rate currently accepted. This discrepancy suggests that some patients with homozygous CBS deficiency may be missed and raises the possibility that there are mildly affected patients who are not diagnosed.106 Further studies in different populations are necessary to clarify this discrepancy.
 Homocystinuria results from AR defects in the CBS gene on chromosome 21q22.3,107 which encodes for CBS, a pyridoxine-dependent hepatic enzyme, which converts homocysteine to cystathionine, a precursor to cysteine. CBS deficiency leads to increased homocysteine levels, and remethylation of this excess homocysteine leads to increased methionine levels.108 Most patients are compound heterozygotes109 with over 92 different mutations reported to date. Particular genotypes show some geographic affinities, with G307S most common in northern Europe (Ireland) and T191M most common in Spanish and South American patients.110 This has some clinical significance as these mutations result in nonpyridoxine-responsive CBS defects.111,112
Homocystinuria results from AR defects in the CBS gene on chromosome 21q22.3,107 which encodes for CBS, a pyridoxine-dependent hepatic enzyme, which converts homocysteine to cystathionine, a precursor to cysteine. CBS deficiency leads to increased homocysteine levels, and remethylation of this excess homocysteine leads to increased methionine levels.108 Most patients are compound heterozygotes109 with over 92 different mutations reported to date. Particular genotypes show some geographic affinities, with G307S most common in northern Europe (Ireland) and T191M most common in Spanish and South American patients.110 This has some clinical significance as these mutations result in nonpyridoxine-responsive CBS defects.111,112
 Elevation of L-homocysteine levels resulting in homocystinuria may also occur due to inherited disorders impairing the biological activation of cobalamin or acquired deficiency of that vitamin, from dietary deficiency or impaired absorption. Additionally, defects resulting in decreased 5-methyltetrahydrofolate may lead to biochemical homocystinuria.
Elevation of L-homocysteine levels resulting in homocystinuria may also occur due to inherited disorders impairing the biological activation of cobalamin or acquired deficiency of that vitamin, from dietary deficiency or impaired absorption. Additionally, defects resulting in decreased 5-methyltetrahydrofolate may lead to biochemical homocystinuria.
 Fibrillin-1 is composed of ∼13% cysteine residues, which are modifiable by homocysteine. Recent studies indicate that homocysteinylation (but not cysteinylation) affects the functional properties of fibrillin-1 (reduced multimerization and fibrillin-1 network deposition) and tropoelastin.113 Thus, the elevated homocysteine levels from CBS deficiency may impair function of fibrillin-1 leading to the phenotypic similarities with Marfan syndrome. Elastic fibers from homocystinuric patients were previously known to show lucent and dense stripes in the elastic matrix and many abnormal elastic microfibrils.32 This could also explain some of the cutaneous features of homocystinuria although the apparent lack of striae in these patients remains unexplained. The mechanism for the neurologic findings is unknown.
Fibrillin-1 is composed of ∼13% cysteine residues, which are modifiable by homocysteine. Recent studies indicate that homocysteinylation (but not cysteinylation) affects the functional properties of fibrillin-1 (reduced multimerization and fibrillin-1 network deposition) and tropoelastin.113 Thus, the elevated homocysteine levels from CBS deficiency may impair function of fibrillin-1 leading to the phenotypic similarities with Marfan syndrome. Elastic fibers from homocystinuric patients were previously known to show lucent and dense stripes in the elastic matrix and many abnormal elastic microfibrils.32 This could also explain some of the cutaneous features of homocystinuria although the apparent lack of striae in these patients remains unexplained. The mechanism for the neurologic findings is unknown.
 Homocystinuria can affect multiple organs, including the skeletal, ocular, vascular, and central nervous systems.114 Most changes are progressive although some manifest at earlier ages than others. Some patients demonstrate involvement of only a single system, such as adult patients who present with a cerebrovascular event.
Homocystinuria can affect multiple organs, including the skeletal, ocular, vascular, and central nervous systems.114 Most changes are progressive although some manifest at earlier ages than others. Some patients demonstrate involvement of only a single system, such as adult patients who present with a cerebrovascular event.
 An asthenic (marfanoid) body habitus is typically present. Additionally, early-onset osteoporosis is prevalent, with up to 50% of patients exhibiting radiographic findings of osteoporosis by age 15.115 Scoliosis, pectus, genu valgum, and a high, arched palate are also common.
An asthenic (marfanoid) body habitus is typically present. Additionally, early-onset osteoporosis is prevalent, with up to 50% of patients exhibiting radiographic findings of osteoporosis by age 15.115 Scoliosis, pectus, genu valgum, and a high, arched palate are also common.
 The typical ocular finding of homocystinuria is ectopia lentis, classically downwardly displaced (differentiating it from the upward displacement seen in Marfan syndrome). Onset is after 1 year of age and it will have developed by 10 years of age in most patients. It may occur earlier in patients who are B6 unresponsive.115 Retinal hemorrhage also occurs.
The typical ocular finding of homocystinuria is ectopia lentis, classically downwardly displaced (differentiating it from the upward displacement seen in Marfan syndrome). Onset is after 1 year of age and it will have developed by 10 years of age in most patients. It may occur earlier in patients who are B6 unresponsive.115 Retinal hemorrhage also occurs.
 The vascular sequelae of homocystinuria can be significant and elevated homocysteine levels in the general population are believed to carry an increased risk for CV morbidity and mortality. Arteriovenous thromboses may occur with emboli affecting the heart, lung, brain, or kidney. Additionally, deep venous thromboses are frequent and can lead to both venous stasis changes and lower extremity ulceration.
The vascular sequelae of homocystinuria can be significant and elevated homocysteine levels in the general population are believed to carry an increased risk for CV morbidity and mortality. Arteriovenous thromboses may occur with emboli affecting the heart, lung, brain, or kidney. Additionally, deep venous thromboses are frequent and can lead to both venous stasis changes and lower extremity ulceration.
 Central nervous system findings exhibit a range of severity. Developmental delay may be the first manifestation of the disease. Mental retardation (reported patients exhibit a range of IQ with B6-responsive patients having a higher IQ on average), seizures, and a variety of psychiatric disorders are described.115
Central nervous system findings exhibit a range of severity. Developmental delay may be the first manifestation of the disease. Mental retardation (reported patients exhibit a range of IQ with B6-responsive patients having a higher IQ on average), seizures, and a variety of psychiatric disorders are described.115
 Cutaneous findings in homocystinuria include prominent malar flushing, which can be exacerbated by vasomotor changes. In one study, malar flushing was more frequently associated with patients who responded to pyridoxine, but the small number of patients makes this finding of unclear significance.114 A thin dermis with prominent appearing vessels is described and livedo reticularis is common. Hair is also described as thin in these patients and pigmentary dilution, due to tyrosinase inhibition from the elevated levels of homocysteine, occurs and may be correctable by appropriate therapy.
Cutaneous findings in homocystinuria include prominent malar flushing, which can be exacerbated by vasomotor changes. In one study, malar flushing was more frequently associated with patients who responded to pyridoxine, but the small number of patients makes this finding of unclear significance.114 A thin dermis with prominent appearing vessels is described and livedo reticularis is common. Hair is also described as thin in these patients and pigmentary dilution, due to tyrosinase inhibition from the elevated levels of homocysteine, occurs and may be correctable by appropriate therapy.
 Marfan syndrome is the main clinical differential diagnosis. Early ectopia lentis may be seen in infants with sulfite oxidase deficiency but otherwise there is not clinical similarity between the conditions.116 Patients with Marfan syndrome or sulfite oxidase deficiency exhibit normal levels of total homocysteine, plasma homocystine, and methionine
Marfan syndrome is the main clinical differential diagnosis. Early ectopia lentis may be seen in infants with sulfite oxidase deficiency but otherwise there is not clinical similarity between the conditions.116 Patients with Marfan syndrome or sulfite oxidase deficiency exhibit normal levels of total homocysteine, plasma homocystine, and methionine
 Several metabolic conditions may exhibit increased homocystine/homocysteine or methionine. Patients with defective metabolism of methionine or its products exhibit elevated methionine levels (but undetectable homocystine and normal/slightly increased homocysteine) and may have methionine adenosyltransferase I/III deficiency, glycine N-methyltransferase deficiency, or S-adenosylhomocysteine hydrolase deficiency.
Several metabolic conditions may exhibit increased homocystine/homocysteine or methionine. Patients with defective metabolism of methionine or its products exhibit elevated methionine levels (but undetectable homocystine and normal/slightly increased homocysteine) and may have methionine adenosyltransferase I/III deficiency, glycine N-methyltransferase deficiency, or S-adenosylhomocysteine hydrolase deficiency.
 Patients with defective methionine remethylation will exhibit increased plasma total homocysteine and homocystine but low levels of methionine. These result from defects in methylenetetrahydrofolate reductase and/or other defects in metabolism of cobalamin. These disorders are often vitamin B12- or folate-dependent. As noted above, defects in metabolism of vitamin B12 resulting from mutations in methylmalonic aciduria and homocystinuria type C protein (MMACHC; the most frequent abnormality of vitamin B12 metabolism) lead to a phenotype of combined methylmalonic aciduria and homocystinuria [cblC; OMIM #277400]. These patients exhibit neurologic, ophthalmologic, and cardiac abnormalities similar to those seen with homocystinuria, probably from the elevated homocysteine.117,118 Findings of cblC typically present in infancy, although late-onset disease is reported, usually characterized by progressive neurologic deterioration. Lastly, secondary hypermethioninemia (no plasma homocystine and normal/mildly increased total homocysteine), can occur due to the hepatic disease caused by tyrosinemia type I or galactosemia, as well as via excessive dietary intake of methionine, such as with high-protein diets or methionine supplemented formula.119
Patients with defective methionine remethylation will exhibit increased plasma total homocysteine and homocystine but low levels of methionine. These result from defects in methylenetetrahydrofolate reductase and/or other defects in metabolism of cobalamin. These disorders are often vitamin B12- or folate-dependent. As noted above, defects in metabolism of vitamin B12 resulting from mutations in methylmalonic aciduria and homocystinuria type C protein (MMACHC; the most frequent abnormality of vitamin B12 metabolism) lead to a phenotype of combined methylmalonic aciduria and homocystinuria [cblC; OMIM #277400]. These patients exhibit neurologic, ophthalmologic, and cardiac abnormalities similar to those seen with homocystinuria, probably from the elevated homocysteine.117,118 Findings of cblC typically present in infancy, although late-onset disease is reported, usually characterized by progressive neurologic deterioration. Lastly, secondary hypermethioninemia (no plasma homocystine and normal/mildly increased total homocysteine), can occur due to the hepatic disease caused by tyrosinemia type I or galactosemia, as well as via excessive dietary intake of methionine, such as with high-protein diets or methionine supplemented formula.119








