Abstract
Lichen planus (LP) is an idiopathic inflammatory disease of the skin and mucous membranes whose primary lesion is a pruritic, violaceous papule. Histologically, a band-like lymphocytic infiltrate underlies an acanthotic epidermis with vacuolar degeneration of the basal cell layer. Several clinically distinct inflammatory dermatoses, including lichen striatus and lichen nitidus, have overlapping histologic features, and as a group they are referred to as lichenoid dermatoses. LP represents a T-cell-mediated autoimmune reaction against epitopes on lesional keratinocytes modified by viral or drug antigens. Clinical variants of LP include annular, atrophic, bullous, hypertrophic, inverse, linear, ulcerative, vulvovaginal–gingival, lichen planopilaris, and drug-induced. Lichen striatus is an asymptomatic, linear dermatosis composed of small flat-topped papules. It resolves spontaneously over months to a few years and primarily affects children. Lichen nitidus is an eruption of multiple, tiny, discrete, shiny papules (often in clusters) that favor the flexor aspects of the upper extremities, the genitalia and the anterior trunk. In erythema dyschromicum perstans, there is a slowly progressive appearance of gray or gray–brown, oval-shaped macules and patches; histologically primarily dermal melanophages are seen.
Keywords
lichen planus, lichenoid dermatoses, lichenoid tissue reaction, lichen striatus, lichen nitidus, lichenoid drug eruption, erythema dyschromicum perstans, keratosis lichenoides chronica, actinic lichen nitidus, annular lichenoid dermatitis (of youth)
Lichen Planus
▪ Lichen ruber planus
- ▪
Idiopathic inflammatory disease of the skin, hair, nails and mucous membranes, seen most commonly in middle-aged adults
- ▪
Flat-topped violaceous papules and plaques favoring the wrists, forearms, genitalia, distal lower extremities, and presacral area
- ▪
Clinical variants include actinic, annular, atrophic, bullous, hypertrophic, inverse, linear, ulcerative, vulvovaginal–gingival, lichen planopilaris, lichen planus pigmentosus, and drug-induced
- ▪
Some lichenoid drug eruptions have a photodistribution, while others are clinically and histologically indistinguishable from idiopathic lichen planus
- ▪
The most commonly incriminated drugs include angiotensin-converting enzyme (ACE) inhibitors, thiazide diuretics, antimalarials, quinidine, and gold
- ▪
Histologically, there is a dense, band-like lymphocytic infiltrate and keratinocyte apoptosis with destruction of the epidermal basal cell layer
- ▪
In this T-cell-mediated autoimmune disorder, basal keratinocytes express altered self-antigens on their surface
Introduction
Lichen planus (LP), the prototype of lichenoid dermatoses, is an idiopathic inflammatory disease of the skin and mucous membranes. Classic LP is characterized by pruritic, violaceous papules that favor the extremities . Histologically, a dense, band-like lymphocytic infiltrate is seen underlying an acanthotic epidermis with hypergranulosis, apoptosis, and destruction of the basal cell layer. The etiology and pathogenesis of LP are not fully understood, but the disorder has been associated with multiple environmental exposures, including viral infections, medications, vaccinations, and dental restorative materials.
LP-like lesions that resemble idiopathic LP may also develop in chronic GVHD, where alloreactive T cells that recognize foreign major histocompatibility complex (MHC) molecules are central effectors (see Ch. 52 ). This lends support to the hypothesis that an autoimmune reaction against epitopes on lesional keratinocytes that have been modified by viral or drug antigens may be responsible for LP.
A number of clinically distinct inflammatory dermatoses have in common varying elements of a lichenoid tissue reaction, and they are referred to as lichenoid dermatoses ( Table 11.1 ).
| MAJOR LICHENOID DERMATOSES AND POSSIBLE ASSOCIATED TARGET ANTIGENS | |
|---|---|
| Lichenoid dermatoses | Possible target antigens |
| Lichen planus | V, D, C, T |
| Lichenoid drug eruption | D |
| Erythema dyschromicum perstans | V, D |
| Graft-versus-host disease (see Ch. 52 ) | Allo, V |
| Keratosis lichenoides chronica | |
| Pityriasis lichenoides * (see Ch. 9 ) | V |
| Lichen nitidus | V |
| Lichen striatus | V |
| Lichen sclerosus (see Ch. 44 ) | V, Auto |
| Fixed drug eruption (see Ch. 21 ) | D |
| Erythema multiforme (see Ch. 20 ) | V, D, C |
| Lupus erythematosus (see Ch. 41 ) | V, Auto, D |
| Dermatomyositis (see Ch. 42 ) | V, Auto, T, D |
| Paraneoplastic pemphigus (see Ch. 29 ) | T |
| Mycosis fungoides (see Ch. 120 ) | T, V |
| Lichenoid pigmented purpura (see Ch. 22 ) | D, V |
| Secondary syphilis (see Ch. 82 ) | |
History
The term lichen planus was initially introduced by Erasmus Wilson in 1869 to describe the condition that had been previously named leichen ruber by Hebra .
Epidemiology
Although its incidence varies depending upon geographic locale, cutaneous LP has been reported to affect from 0.2% to 1% of the adult population , whereas oral lesions have been observed in up to 1–4% of the population. There is no overt racial predisposition. LP most commonly has its onset during the fifth or sixth decade, with two-thirds of patients developing the disease between the ages of 30 and 60 years. It is rare in both infants and the elderly and, typically, only 1–4% of patients are children. However, more recent studies have suggested that LP may actually be more common in children, especially in Arab populations. Oral LP is also quite uncommon in young people and it usually affects middle-aged to elderly individuals (mean age at diagnosis being 52 years). Although LP is frequently thought to have no gender predilection, some studies have found that women were affected nearly twice as often as men.
Mucosal involvement, particularly oral lesions, is observed in up to 75% of patients with cutaneous LP, but the former can be the only manifestation of the disease. Only 10–20% of patients whose initial presentation is oral LP will eventually develop cutaneous LP. Although reports of familial LP are rare, it may occur more frequently than previously thought; for example, LP occurs in up to 10% of first-degree relatives of affected patients. Cases of familial LP tend to have an earlier age of onset, a higher relapse rate, and more frequent oral mucosal involvement. Of note, reports of concurrent LP in monozygotic twins who were living together suggest an environmental trigger.
Pathogenesis
There is a growing body of evidence that LP represents T-cell-mediated autoimmune damage to basal keratinocytes that express altered self-antigens on their surface.
Target antigens
Clinical observations and anecdotal evidence have long suggested a relationship between exposure to a number of exogenous agents (e.g. viruses, medications, contact allergens) and the development of LP (see Table 11.1 ). In theory, T cells that normally do not respond to skin (epidermis)-restricted antigens are primed by exogenous agents when the latter possess cross-reactive antigens that can activate autoreactive T cells via molecular mimicry. In other words, the T-cell receptors on the T cells that induce mucocutaneous LP could cross-react with exogenous antigens.
Hepatitis C virus
Of the many potential exogenous antigens, significant attention has been focused on the possible role of viruses, particularly hepatitis C virus (HCV). In several case–control studies, the prevalence of HCV (3.5–38%) was 2- to 13.5-fold higher in patients with LP than in controls. This association seems to be strongest in Japanese and Mediterranean populations, probably due to the high prevalence of HCV infection in these countries. In the US, one case–control study found that 12 (55%) of 22 patients with LP had anti-HCV antibodies, and this was significantly higher than the 25% of 40 psoriatic patients or the 0.17% of blood donors who tested positive . A more recent systematic review and meta-analysis of existing epidemiologic studies demonstrated that an association between LP and HCV infection did exist in certain geographic regions (e.g. East and Southeast Asia, South America, the Middle East, Europe), but not in others (e.g. North America, South Asia, and Africa) .
Of the various types of LP, it is the oral form that is most commonly viewed as a manifestation of HCV infection. By PCR, HCV RNA was detected in 93% of oral LP lesions , suggesting HCV replication within LP lesions. However, these PCR results were not confirmed by other studies.
Of note, an increased frequency of the HLA-DR6 allele has been reported in Italian patients with HCV-associated oral LP , raising the possibility that CD4 + T cells activated upon recognition of HCV-encoded peptides bound to HLA-DR6 molecules could be directly involved in the pathogenesis of LP. In support of this possibility, HCV tetramer analysis has shown that HCV-specific CD4 + and/or CD8 + T cells are present with higher frequency in oral LP lesions compared with the circulating compartment, suggesting that they play a role in the pathogenesis of LP .
In patients receiving anti-HCV therapy, originally interferon and ribavirin and more recently protease and polymerase inhibitors, the effects on mucocutaneous LP have varied . Lesions have improved in some patients while others experienced a flare of disease activity.
Other viruses
With regard to the role of other viruses in LP, human herpesvirus (HHV)-6 was detected in 65–100% of oral LP lesions by in situ hybridization and immunohistochemical techniques when it was absent from normal oral tissues. In addition, a retrospective survey of 18 lesional LP tissue samples (as well as 11 non-lesional LP and 11 lesional psoriasis samples) found that 11 of the 18 lesional LP samples contained HHV-7 DNA as compared to 1 of 11 and 2 of 11 non-lesional LP and lesional psoriasis samples, respectively . Remission of LP was associated with decreased HHV-7 protein expression, in particular within infiltrating plasmacytoid dendritic cells .
There are also sporadic case reports of LP lesions developing in areas recently affected by herpes simplex virus ( HSV ) or varicella–zoster virus ( VZV ) infections (see Table 80.7 ). Possible explanations include a nonspecific Koebner phenomenon or isotopic response or an immune reaction to the virus. In a recent immunohistochemical study involving eight patients, VZV-gE antigen was detected within the eccrine epithelia of LP lesions if they were in a dermatomal (zosteriform) pattern but not if they were in a linear array along the lines of Blaschko . Two of the patients with the dermatomal pattern reported no preceding episode of herpes zoster, leading the authors to speculate if LP could be triggered by subclinical VZV reactivation. A link between human papillomavirus ( HPV ) and oral LP has also been suggested based upon the identification of a clonal expansion of HPV-16-specific CD8 + T cells within the lesions .
While measurements (via PCR) of viral genomes within lesional skin or blood have been inconclusive, as noted above, virus-specific T cells have been detected within lesions. This suggests that the pathology is a direct consequence of immune responses, perhaps to virally induced alterations in the antigenicity of epidermal cells rather than the viruses themselves. However, it is also possible that virus-specific T cells are nonspecifically trapped within sites of inflammation, having been activated systemically or locally at distant sites, and then they expand and mediate tissue damage “accidently” due to cross-reactivity with other antigens (e.g. drugs) .
Vaccines
A number of reports have described the appearance of LP after administration of influenza vaccines and different types of HBV vaccines . The time interval between the initial dose and the development of cutaneous or mucosal lesions has varied from a few days to 5 months. One recommendation is that patients who develop LP before completing their vaccination series should avoid further injections because of an increased risk of developing severe LP lesions, including the bullous variant.
Bacteria
Investigations regarding a relationship between bacteria and LP have been limited. In particular, a definitive etiologic role for Helicobacter pylori has not been proven.
Contact allergens
The role of contact allergy to a variety of metals in the exacerbation or induction of oral LP has been well described, based on exposure to metallic dental restorations or constructions, positive patch test results, and then regression or complete clearing after removal of the sensitizing metal and replacement with other materials. Because involved allergens are dissolved and spread via saliva, mucosal reactions may extend beyond the contact areas. The metals that aggravate oral LP include amalgam (mercury), copper, and gold. Although approximately 95% of patients had improvement after removal of the sensitizing metal, 75% of patients with negative patch test results also reported clearing of oral LP after removal of the metal and replacement with other materials. One possible explanation for this finding is that mercury served as an irritant and induced lesions via the Koebner phenomenon.
Of note, the development of contact allergy to metals within dental restorations in patients with LP could be explained by easy penetration of the metal via damaged mucosa.
Drugs
Cutaneous eruptions similar or even identical to LP, both clinically and histologically, have been linked to a variety of drugs. The terms “lichen planus-like” and “lichenoid” are often used to describe this phenomenon . A wide variety of drugs have been associated with lichenoid drug eruptions and the list of such drugs steadily increases ( Table 11.2 ). However, recurrence of the lesions subsequent to drug rechallenge has not been documented for the majority of these drugs.
| DRUGS IMPLICATED IN LICHENOID DRUG ERUPTIONS | |
|---|---|
| Antimicrobials | |
|
|
| Antihypertensives | |
|
|
| Antimalarials | |
|
|
| Antidepressants, anti-anxiety drugs, antipsychotics and anticonvulsants | |
|
|
| TNF-α inhibitors | |
|
|
| Diuretics | |
|
|
| Hypoglycemic agents | |
|
|
| Metals | |
|
|
| NSAIDs | |
|
|
| Miscellaneous drugs | |
|
|
* Also used to treat hypoglycemia.
Autoantigens, including tumor antigens
In occasional patients, LP has been reported as a possible autoimmune reaction triggered by an underlying neoplasm. Also, a lichenoid tissue reaction is seen in patients with paraneoplastic pemphigus. The temporal relationship between LP and the underlying neoplasm in the reported patients suggests that the neoplasms may have stimulated a cell-mediated immune response against tumor antigens that led to the generation of autoreactive T cells that cross-reacted against antigens expressed on epidermal cells.
Although numerous case reports describe patients with both LP and autoimmune diseases, studies with larger numbers of patients with LP have shown no increased incidence of autoimmune diseases. Multiple investigators have described a significant association between specific HLA antigens and LP; for example, an increased frequency of HLA-B27, HLA-B51, HLA-Bw57 (oral LP in English patients), HLA-DR1 (cutaneous and oral LP), HLA-DR9 (oral LP in Japanese and Chinese patients), and HLA-DR6 (HCV-associated oral; see above). However, a true association with a particular HLA allele has been difficult to establish because of significant geographic heterogeneity and clinical patient selection.
A murine model of LP has been established by employing autoreactive T cells capable of producing interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) ( Fig. 11.1 ) . Intradermal inoculation of CD4 + autoreactive T-cell clones into the footpads of syngeneic mice can induce local histologic changes similar to LP or lichenoid skin diseases. In this model, the autoreactive T cells can respond to self MHC class II antigens constitutively expressed on macrophages and Langerhans cells, and they migrate into the epidermis, resulting in epidermal injury. These T cells, therefore, can induce LP-like lesions without any alteration in the antigenicity of the epidermis. In subsequent studies, desmoglein-specific T cells were also capable of inducing LP-like changes histologically . Thus, LP could be induced by different types of T cells – those that specifically target antigens constitutively expressed in the epidermis but also T cells that target antigens not expressed within the epidermis.
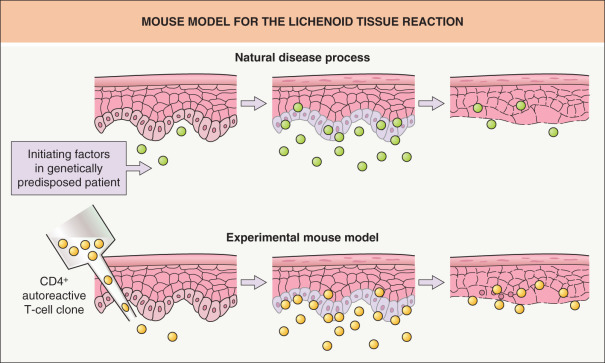
Additionally, in the natural disease process, exogenous agents (e.g. viral infections, drugs) could induce alterations in the antigenicity of epidermal cells and trigger the activation of T cells. Of note, such autoaggressive reactions could function to eliminate abnormal keratinocytes altered by these exogenous agents. However, in the situation where T cells initially responding to self-antigens modified by exogenous agents subsequently become cross-reactive with some self-epitopes, these T cells would then chronically respond to the previously ignored self-epitopes, leading to perpetuation of an autoimmune attack by such T cells rather than elimination of the abnormal keratinocytes.
Effector cells
There are conflicting data regarding the phenotype of the inflammatory infiltrate in LP lesions. Although initial immunohistochemical studies showed that the cellular infiltrate contained an increased ratio of CD4 + : CD8 + T cells , other investigators found a predominance of CD8 + T cells, particularly in older lesions. Evidence to support the crucial role of CD8 + T cells in autoimmune damage to basal keratinocytes has been provided by CD8 + T cells isolated from lesional skin; these T cells exhibited specific cytotoxic activity against autologous lesional and normal keratinocytes .
Basal cell damage as evidenced by apoptotic DNA fragments is greatest in the epidermis. A possible mechanism is as follows: IFN-γ produced by CD8 + T cells upregulates Fas expression by keratinocytes, rendering them susceptible to T-cell-mediated, Fas ligand-driven apoptosis. This interaction triggers a cascade of intracellular enzymatic reactions resulting in DNA fragmentation (see Ch. 107 ). In addition to Fas, death receptor-induced apoptosis involves signaling processes via TNF-R1, TRAIL-R1 and 2, and DR3 or DR6. Because type 1 helper T cells (Th1), such as the autoreactive CD4 + T cells in the murine model (see Fig. 11.1 ), can produce large amounts of IFN-γ and TNF-β upon activation and thereby induce or enhance the expression of apoptosis-associated proteins such as Fas and TRAIL, they may also play a role in extensive epidermal damage by promoting apoptotic death of keratinocytes. More recent studies have shown that granule exocytosis with release of perforin and granzyme B, rather than the Fas/Fas-ligand system, is the main pathway of cytotoxicity mediated by CD4 + and CD8 + T cells in humans . However, a combination of the two mechanisms is most likely, with predominance depending upon the particular stage in the disease process.
The innate immune system, regulatory T cells (Tregs), and Th17 cells
Recent studies have provided evidence for the involvement of Toll-like receptor (TLR) signaling in the induction of autoimmunity. As depicted in Fig. 4.1 , certain TLRs recognize viral RNA as well as imidazoquinolones (e.g. imiquimod). Interestingly, topical application of imiquimod, which can lead to enhanced migration and maturation of dermal dendritic cells, followed by increased production of proinflammatory cytokines and activation of antigen-specific CD8 + T cells, may exacerbate lesions of LP.
In some autoimmune diseases, T-cell differentiation may shift from Treg cells (immune suppression) to Th17 cells (see Ch. 4 ), but an increase in both populations has been found in LP lesions. However, the functionality of the Treg cells within these lesions has been questioned.
Effector T cells’ access to the epidermis
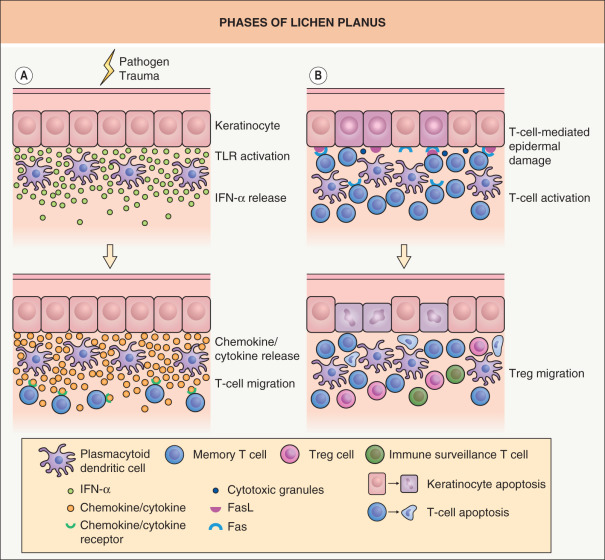
A critical event in the initiation of immune responses in LP lesions is for memory T cells to migrate from the circulation into a particular skin site. Release of type 1 IFNs, such as IFN-α, from activated plasmacytoid dendritic cells (pDCs) and keratinocytes may be critical in inducing skin-directed migration of effector memory T cells ( Fig. 11.2A ). Stimulation of TLRs expressed on pDCs and keratinocytes by pathogens or endogenous ligands (released via skin damage) represents one of the earliest events and it is sufficient to induce type 1 IFN production. Type 1 IFN signaling and type 1 IFN-inducible chemokines (e.g. IP-10/CXCL10) then serve to recruit chemokine receptor CXCR3-expressing effector memory T cells (Th1 cells) into the skin via CXCR3/IP-10 interactions ( Table 11.3 ). A number of other molecular interactions, such as CCR4/TARC, CCR10/CTACK and LFA-1/ICAM-1 expressed on T cells and keratinocytes, respectively, have also been implicated in the recruitment of memory T cells and pDCs to the dermal–epidermal junction . Because IFN-α from pDCs induces IFN-γ production by T cells and IFN-γ also sustains IFN-α production, a positive feedback loop may be operational within LP lesions. This ordered sequence of events provides a possible explanation for why LP lesions develop within traumatized sites and virally induced lesions.
| CHEMOKINES AND CHEMOKINE RECEPTORS INVOLVED IN T-CELL MIGRATION INTO THE SKIN | ||
|---|---|---|
| Chemokines * | Chemokine receptors | Functional type of chemokines/T cells attracted † |
| RANTES (CCL5) MCP-2 (CCL8) Eotaxin-1, -2, -3 (CCL11, 24, 26) | CCR3 | Inflammatory/Th2 cells |
| TARC (CCL17) MDC (CCL22) | CCR4 | Inflammatory/Th2 cells |
| MIP-1α,β (CCL3, 4) RANTES (CCL5) MCP-2 (CCL8) | CCR5 | Inflammatory/Th2 cells |
| CCL1 | CCR8 | Homeostatic/memory T cells |
| CTACK (CCL27) MEC (CCL28) | CCR10 | Homeostatic/memory T cells |
| MIG (CXCL9) IP-10 (CXCL10) I-TAC (CXCL11) | CXCR3 | Inflammatory/Th1 cells |
| CXCL16 | CXCR6 | Inflammatory/Th1 cells |
* Cytokines that have chemoattractant activity; two major groups are differentiated based on the position of two cysteine (C) residues compared with the other amino acid residues (X): CXC- or α-chemokines and CC- or β-chemokines.
† “Inflammatory” chemokines are produced at sites of cutaneous inflammation and mediate skin-directed migration of memory T cells (Th1, Th2); “homeostatic” chemokines are constitutively produced within non-inflamed skin and mediate skin-directed migration of memory T cells with an “immune surveillance” function.
Although chemokines produced at inflammatory skin sites are thought to regulate the composition of the Th1- or Th2-driven cellular infiltrates, memory T cells with a “surveillance” function can also migrate to skin sites under non-inflamed conditions ( Fig. 11.2B ). “Homeostatic” chemokines constitutively produced under non-inflamed conditions can mediate the skin-directed migration of these “immune surveillance” T cells (see Table 11.3 ), leading to clearance of invading pathogens such as viruses. In addition, Treg cells that have the capacity to suppress activated T cells are also able to enter inflamed skin sites. However, in LP lesions, there is no definitive means of distinguishing “immune surveillance” T cells or protective Treg cells from “pathogenic” T cells. Lastly, identification of the cells responsible for LP is further complicated by the presence of both skin tissue-resident memory T cells (TRM cells) and migratory central memory T cells (TCM cells) within skin lesions, with the former, whose physiologic role includes local protection against pathogens, becoming a mediator of tissue damage.
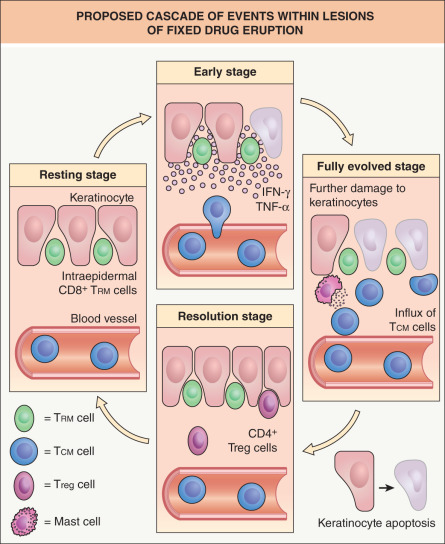
Of note, while the epidermotropic migration of effector T cells leading to epidermal damage in lichenoid tissue reactions is clearly a complicated, multistep process, the latter can be bypassed (at least in part) in the case of fixed drug eruptions (FDE). This is because effector CD8 + T cells responsible for the epidermal damage persist as a stable population within previously affected sites, even when the skin becomes normal-appearing . In particular, CD8 + TRM cells (see above) populate the epidermis of “resting” FDE lesions and although these cells are the principal mediators of localized tissue damage, CD4 + and CD8 + TCM cells, recruited from the circulation, also contribute to the epidermal damage ( Fig. 11.3 ).
Sweating disturbance
Sweat contains interleukin (IL)-1, IL-6, IL-8, and TNF-α, proinflammatory cytokines that have been implicated in the induction of T-cell recruitment into the skin . In theory, extravasated sweat could explain the syringotropic T-cell migration occasionally seen in the early lesions of LP. Whether leakage of sweat near the dermal–epidermal junction also represents an early event in the inflammatory cascade remains to be determined.
Clinical Features
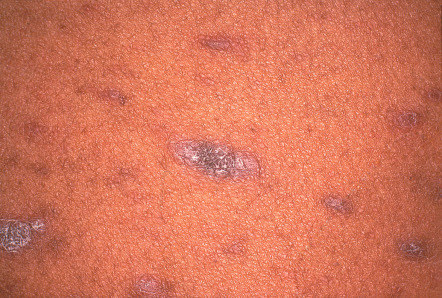
The characteristic primary lesion of LP is a small, polygonal-shaped, violaceous, flat-topped papule ( Fig. 11.4 ); occasionally papules are umbilicated. The surface is slightly shiny or transparent, and a network of fine white lines called “Wickham striae” or small gray–white puncta are also seen. The latter correspond histologically to focal thickening of the granular layer. Wickham striae are readily apparent by dermoscopy (see Ch. 0 ). Postinflammatory hyperpigmentation is also a common finding ( Fig. 11.5 ).
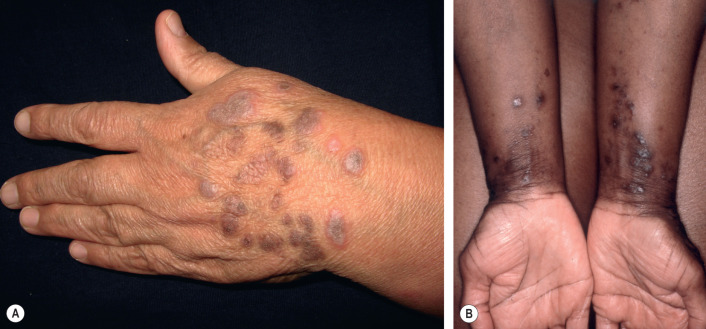
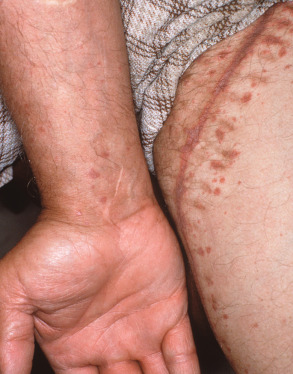
The papules of LP may be widely dispersed or they may cluster or coalesce into larger plaques. LP is usually pruritic. Although the Koebner phenomenon (i.e. isomorphic response) is commonly seen in LP ( Fig. 11.6 ), excoriations and impetiginization are unusual. A linear array of lesions can be seen as a consequence of the Koebner phenomenon or an isotopic response at the site of healed herpes zoster, as well as in the linear variant of LP which follows the lines of Blaschko.
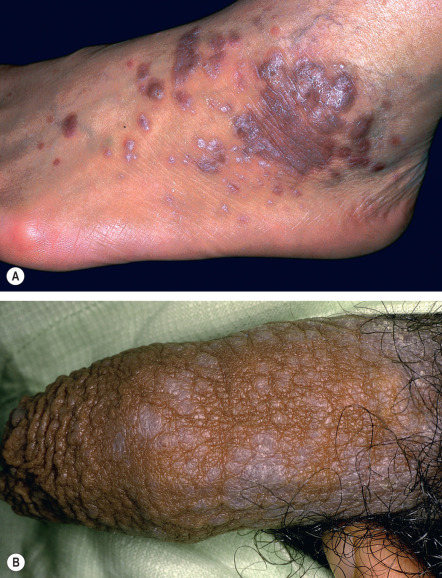
The most common sites of involvement are the flexor wrists and forearms, the dorsal hands, the shins, and the presacral area. Mucous membranes, especially the oral mucosa (see below), are affected in more than half of patients, and this is often the only site of disease. Lesions are also commonly seen on the glans penis ( Fig. 11.7A ), where they can have an annular or figurate configuration and may become erosive. Several of the distinctive clinical variants of LP are discussed separately.
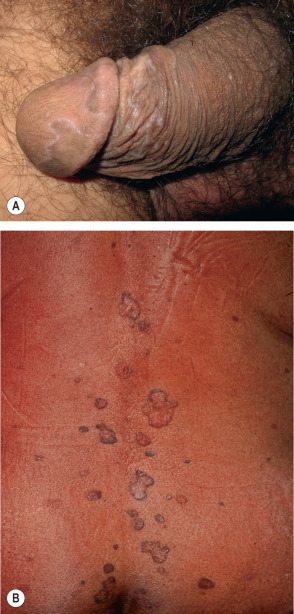
The duration of the disease can vary depending upon the LP variant. While the lesions of exanthematous LP typically resolve within a year, hypertrophic, oral and nail LP tend to be more persistent. In particular, ulcerative oral LP may be a lifelong affliction.
Actinic LP
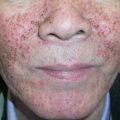
This variant is reported under a variety of names, including LP actinicus, LP subtropicus, LP tropicus and lichenoid melanodermatitis. Although the majority of reported patients have been from Middle Eastern countries, this variant has been observed worldwide. Most patients are young adults or children, but there is no gender predilection. Onset of this variant is typically during the spring and summer, and lesions primarily involve sun-exposed skin of the face, followed by the neck and dorsal surfaces of the hands and arms. The lesions usually consist of red–brown plaques with an annular configuration, but melasma-like hyperpigmented patches have been observed. In temperate climates, spontaneous improvement may occur during the winter months.
Acute (exanthematous) LP
Because lesions are usually widely distributed and disseminate rapidly, this form is also known as exanthematous or eruptive LP. The commonly affected areas include the trunk ( Fig. 11.8 ), the inner aspects of the wrists and the dorsal feet. Reports in the literature of this variant probably include lichenoid drug eruptions. The clinical course is usually self-limited, and in general lesions resolve with hyperpigmentation within 3 to 9 months.