Inflammatory Diseases that Simulate Lymphomas: Cutaneous Pseudolymphomas: Introduction
|
Cutaneous pseudolymphoma is a term used to describe skin lesions that bear a clinical and/or histopathologic resemblance to lymphoma. Over the years, a wide variety of designations have been included in this category. Some of these represent outdated synonyms, some refer to variants of the same entity, and some are misnomers for diseases now recognized to be true lymphomas. The relevant entities that are discussed in this and other chapters can be organized into several distinct disorders based on their lymphoid subset composition, pattern of cutaneous infiltration, and associated clinical findings (Table 146-1).
Clinicopathologic Subtype | Predominant Lymphoid Subset(s) | Predominant Localization | Major Associated Findings |
|---|---|---|---|
Cutaneous lymphoid hyperplasia | B and T cell | Reticular dermis | — |
Kimura disease | B and T cell | Subcutis | Lymphadenopathy |
Angiolymphoid hyperplasia with eosinophilia | B and T cell | Reticular dermis | Eosinophilia |
Castleman disease | B and T cell | Subcutis | Lymphadenopathy, POEMS syndrome |
Pseudomycosis fungoides | T cell | Papillary dermis and epidermis | — |
Lymphomatoid contact dermatitis | T cell | Papillary dermis and epidermis | Contact allergen |
Lymphocytic infiltration of the skin (Jessner’s) | T cell | Perivascular and periadnexal dermis | — |
Cutaneous Lymphoid Hyperplasia
Cutaneous lymphoid hyperplasia (CLH) has a worldwide distribution and affects all races and ethnic groups. It occurs in both adults and children. Females are more commonly affected than males.
CLH is characterized by a relatively dense lymphoid infiltrate, centered in the reticular dermis, which is usually B-cell rich and may resemble lymphoma clinically and/or histopathologically. Many terms are used to refer to this type of pseudolymphoma, including Spiegler–Fendt sarcoid, lymphocytoma cutis, lymphadenosis benigna cutis, and cutaneous lymphoplasia. Cutaneous lymphoid hyperplasia is the preferred term because it accurately describes the underlying pathophysiology of the lesion and is unlikely to be confused with terms used to describe various forms of cutaneous lymphoma.1–6
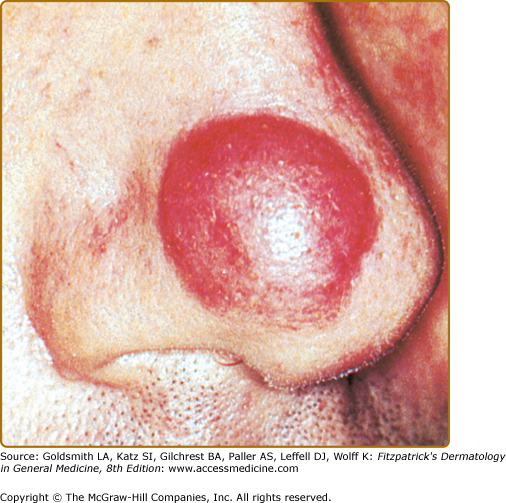
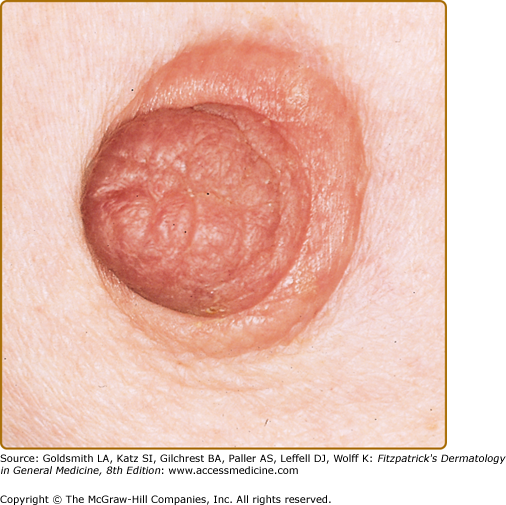
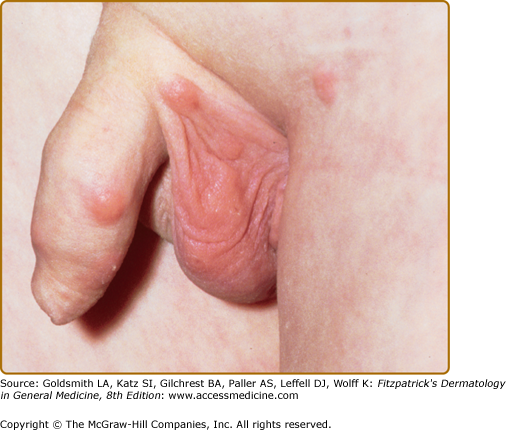
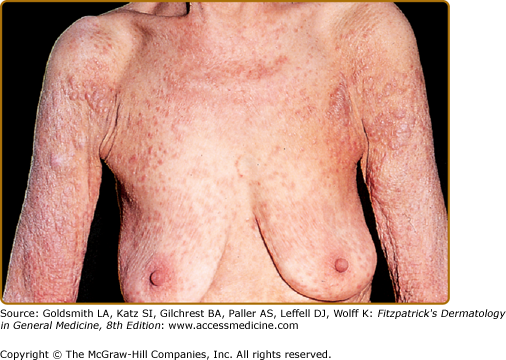
In most cases, CLH is idiopathic; however, some lesions are associated with exposure to foreign antigens from arthropods (bites, stings, infestations), infections (herpes zoster, Borrelia burgdorferi, Helicobacter pylori), tattoos, acupuncture, trauma, gold jewelry, vaccinations, hyposensitization injections, or medications (Figs. 146-1, 146-2, 146-3, and 146-4).1–9 Medications that may induce CLH include phenytoin, carbamazepine, phenobarbital, β blockers, calcium channel blockers, angiotensin-converting enzyme inhibitors, allopurinol, d–penicillamine, penicillin, mexiletine chloride, cyclosporine, and agents that inhibit binding of histamine to H1, H2, or H1c receptors. These drugs include conventional H1 and H2 antagonists, as well as tricyclic and nontricyclic antidepressants and phenothiazines.10 It has been proposed that, rather than being the target of an immune response themselves, these latter agents may alter lymphocyte reactivity in a way that promotes a CLH response to other antigens in some individuals.11,12
Although the details of the workup for individual patients will vary with the specific type of pseudolymphoma being considered, certain data are generally important for establishing the correct diagnosis, identifying potential causative factors, and ruling out an underlying lymphoma. The clinical history taking should elicit information about the duration and symptomatology of lesions; the nature and pace of clinical progression; past treatment; local or systemic exposure to foreign antigens, including medications; and personal or family history of other lymphoproliferative disorders. It should also include a review of systems focusing on so-called lymphoma B symptoms, such as fever of unknown origin, unexplained weight loss, night sweats, fatigue, and malaise. A general physical examination is important, with special attention to the type and distribution of skin lesions and to the status of peripheral lymph nodes, liver, and spleen. A complete blood count with differential review, general chemistry panel, and a chest radiograph are screening tests that help to exclude extracutaneous involvement. Lesional skin biopsy is an essential part of the diagnostic evaluation. The use of topical and systemic glucocorticoids should be discontinued approximately 4 weeks before biopsy if possible, because these agents can attenuate lymphoid infiltrates and thereby confound their interpretation. Skin specimens should be large enough and deep enough to ensure representative sampling and to provide adequate tissue for routine histopathologic, immunohistologic, and molecular biologic studies. As with squamoproliferative skin lesions, the most diagnostic areas within cutaneous lymphoid infiltrates are often their deepest portions (Fig. 146-5). For cost-effectiveness, one can often perform these biopsy studies in a sequential fashion, proceeding to the next test only if a firm diagnosis cannot be made based on the results of preceding analyses. However, rigorous characterization of a pseudolymphomatous infiltrate requires the full complement of these assays. If there is only a solitary lesion, it is generally best to obtain all biopsy material at one time, because inflammatory changes induced by the initial biopsy may interfere with the interpretation of subsequent specimens from the same locale.
In the event that there is still uncertainty as to whether the lymphoid infiltrate represents a pseudolymphoma or a lymphoma, additional workup can be helpful. This includes computed tomographic scans of the chest, abdomen, and pelvis, as well as bone marrow aspiration and biopsy. Biopsy can be performed on abnormally enlarged lymph nodes identified by physical examination or radiography and the specimens evaluated as described in the preceding paragraph for skin lesions. It is possible for patients with atypical cutaneous B-cell infiltrates manifesting as only one or a few skin lesions to have an otherwise unremarkable workup until radiographic scans of deep lymph nodes or bone marrow sampling demonstrates advanced-stage B-cell lymphoma with minor skin involvement.
There may be a history of contact with an etiologically relevant foreign antigen; however, the majority of CLH cases are idiopathic. Lesions may be pruritic or asymptomatic. Occasionally, they may be slightly tender.13
CLH presents most commonly as a solitary nodule, but it can also appear as a localized array of nodules, plaques, or papules. Generalized forms occur rarely. The head, neck, extremities, breasts, and genitalia are common sites (see Figs. 146-1, 146-2, 146-3, and 146-4). Lesions have a doughy to firm consistency and range from red–brown to violaceous in color. Lesions secondary to arthropod bites can be pruritic, ulcerated, or crusted. Occasionally, reactive lymphadenopathy may be present in the area of regional lymphatic drainage.13,14
Hydantoin-associated pseudolymphoma syndrome is caused by anticonvulsant drugs such as phenytoin. This syndrome is characterized by fever, lymphadenopathy, hepatosplenomegaly, arthralgia, eosinophilia, and generalized cutaneous macules and papules or, rarely, nodules.4,6 Nodules may contain aggregates of large lymphoid cells that raise concern for lymphoma; however, both B and T cells are polyclonal. Subcutaneous lymph nodes may exhibit reactive follicular hyperplasia or atypical features suggestive of lymphoma. The macular and papular lesions of this syndrome share histopathologic features with those of hypersensitivity reactions to other drugs.
Acral pseudolymphomatous angiokeratoma of children presents as a unilateral eruption of angiomatous papules on the extremities.15 There is a dense lymphoid infiltrate associated with histiocytes, plasma cells, and prominent, thickened capillaries.16 This disorder is probably a variant of CLH secondary to arthropod bites.
Large cell lymphocytoma, originally reported as a cutaneous pseudolymphoma, most likely represents a mixed cell or large cell form of primary cutaneous B-cell lymphoma (CBCL).5
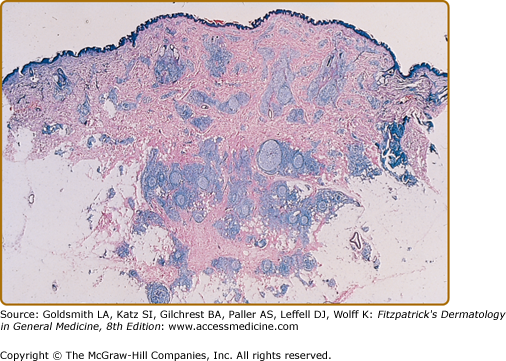
Histopathologic examination of CLH lesions reveals a dense, nodular or diffuse lymphoid infiltrate that is concentrated in the reticular dermis (see Figs. 146-5 and 146-6). It tends to be top heavy and to taper out in the lower dermis, although more widespread dermal infiltration with extension into the subcutis can sometimes be seen. In most cases, the epidermis is normal and separated from the underlying infiltrate by a narrow grenz zone of uninvolved papillary dermis. Occasionally, the epidermis may exhibit variable degrees of hyperkeratosis, parakeratosis, acanthosis, dyskeratosis, spongiosis, and basal vacuolar degeneration. Arthropod-induced reactions may show prominent epidermal hyperplasia or ulceration. Rarely, retained mouth parts may be seen associated with foreign-body granulomatous reactions. The dermal infiltrate is composed primarily of small mature lymphocytes, with a minor component of large lymphoid cells containing large, pale vesicular nuclei with small nucleoli (Fig. 146-7). Fewer than half of cases exhibit well-defined reactive lymphoid follicles discernible in routinely stained sections (see Figs. 146-5 and 146-8). These B-cell follicles may be either primary or secondary. The former are composed of homogeneous small lymphocytes, whereas the latter are compartmentalized into a peripheral mantle zone of small lymphocytes surrounding a germinal center composed of a heterogeneous mixture of small and large lymphoid cells with cleaved and noncleaved nuclei known as centrocytes and centroblasts. Germinal centers contain CD4+ T-follicular helper cells characterized by expression of PD-1, Bcl-6, and CXCL13.17 Also present in germinal centers are tingible-body macrophages, so named because they contain phagocytized debris from apoptotic lymphoid cells. Mitotic figures may be numerous within germinal centers but are usually uncommon elsewhere in the infiltrate. Various types of histiocytes, including macrophages, dermal dendritic cells, and Langerhans cells, are scattered throughout the infiltrate in variable proportions. Capillary hyperplasia and endothelial swelling are often present. Other cells are sometimes admixed, including plasma cells, eosinophils, mast cells, neutrophils, and histiocytic giant cells. The first two are particularly common in arthropod-induced reactions such as nodular scabies (see Fig. 146-6).13,14
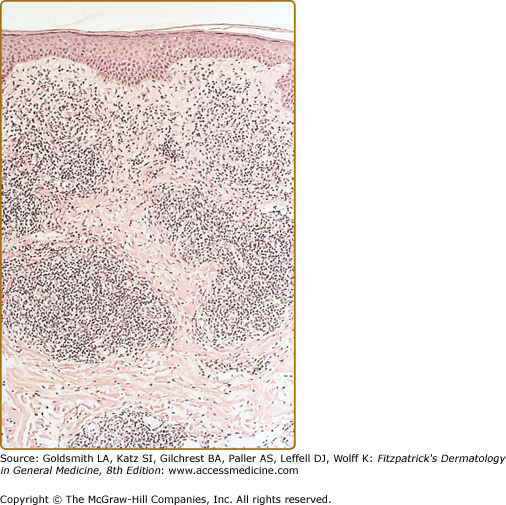
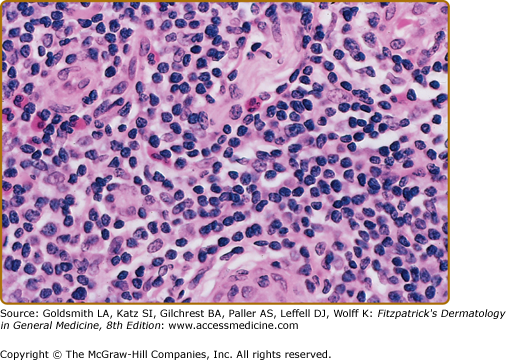
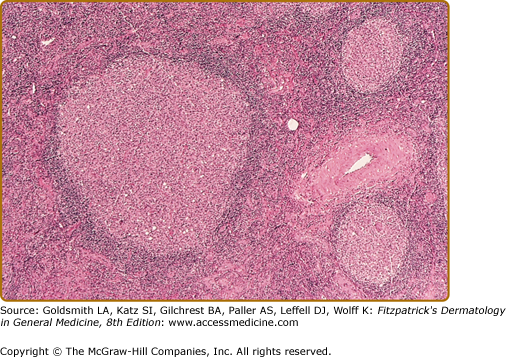
In the less common T-cell CLH, there are only rare B cells.4–6,18–20 Lesions may be idiopathic or secondary to drugs or arthropod bites. Sparse to moderate epidermotropism by atypical lymphoid cells may be seen. The dermal infiltrate has a nodular or diffuse pattern and may contain cerebriform cells, immunoblasts, and histiocytes mixed with a preponderance of small mature lymphocytes. The histiocytes are sometimes numerous enough to impart a vaguely granulomatous appearance.18 The author has seen one such case presenting as multiple nodules on the trunk and extremities that was associated with rheumatoid arthritis and eventuated in T-cell lymphoma.
CLH is just one special case of extranodal lymphoid hyperplasia that may occur in a variety of organ systems. In their most well-developed form, these extranodal reactions recreate the immunoarchitectural features of reactive follicular lymphoid hyperplasia occurring in lymphoid tissues. CLH is generally a mixed B-cell/T-cell disorder. B cells may be organized into primary or secondary lymphoid follicles, simple clusters devoid of the follicular dendritic cell network seen in B-cell follicles, or may be randomly scattered throughout the infiltrate (Fig. 146-9). A defining immunophenotypic feature of CLH is that the small, nongerminal center B cells and plasma cells are polytypic; i.e., instead of restriction to only one type of immunoglobulin light chain, there is a mixture of κ-positive and λ-positive cells.21–23 The follicular dendritic cell networks in lymphoid follicles coexpress both immunoglobulin light chains because of the polytypic nature of the immunoglobulins bound to these networks (Fig. 146-10). The larger germinal center B cells are also polytypic but generally have very low levels of immunoglobulin expression, which is obscured by the more intense staining of the follicular dendritic cell networks in which they are enmeshed.24 Consistent with their role as a site of antigen selection and apoptosis, reactive germinal centers test negative for the bcl-2 oncoprotein.5
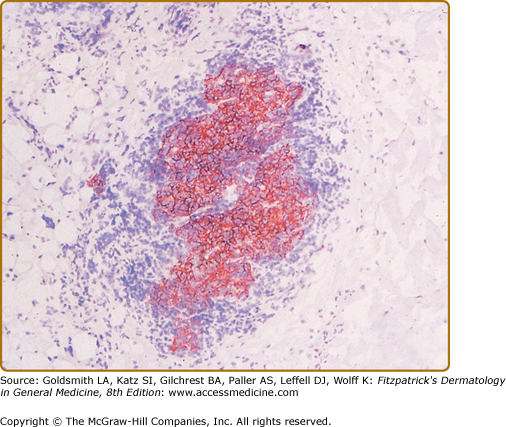
Figure 146-10
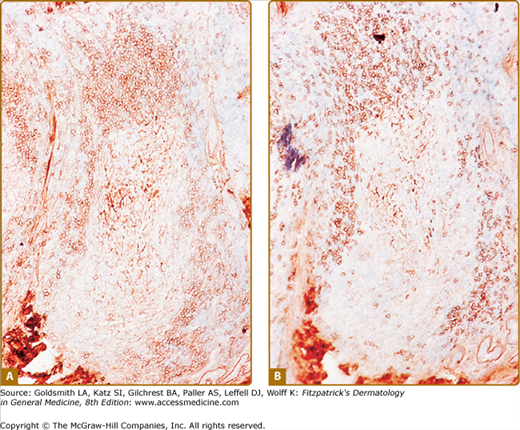
Cutaneous lymphoid hyperplasia. Reactive lymphoid follicle contains a germinal center surrounded by a mantle zone composed of a mixture of B cells that express either κ-immunoglobulin light chain (A) or λ-immunoglobulin light chain (B). Note that the central follicular dendritic cell network stains positive for both light chains because polytypic immunoglobulins are bound to its surface.
Some researchers classify CLH into follicular and diffuse variants based on the identification of lymphoid follicles in routinely stained sections. This distinction is probably not relevant biologically, because most of the apparently diffuse cases also exhibit B-cell follicles when examined immunohistologically. T cells are also present in all cases of CLH. They are found in small numbers within lymphoid follicles, for example, T follicular helper cells, but are concentrated mainly between follicles, constituting an interfollicular T-cell domain. This imparts an overall compartmentalization of B and T cells in CLH lesions. The CD4+ T-cell subset predominates over the CD8+ subset. There is a normal pattern of T-cell antigen expression except that CD7 may be moderately deficient at times, being expressed by only 20% to 40% of T cells. Germinal center B cells, mantle zone B cells, interfollicular B cells, and plasma cells also express the same complement of cell surface differentiation antigens expressed by their counterparts in reactive lymphoid tissues.
Unlike in most nodal follicular B-cell lymphomas (Chapter 145), the t(14;18) translocation, which results in inappropriate expression of the bcl-2 oncoprotein, is absent in CLH.5,25–27 Molecular biologic studies show that 5% to 33% of CLH cases defined by clinicopathologic and immunohistologic criteria contain occult dominant immunoglobulin gene rearrangements, which indicate the presence of a dominant B-cell clone.5,28–32 These cases, known as clonal CLH, have progressed in some cases to overt B-cell lymphoma exhibiting the same dominant B-cell clone. These findings support the view that CLH, clonal CLH, and primary CBCL exist as points along a continuum of cutaneous B-cell lymphoproliferative disorders. Progression across this spectrum of disease clearly occurs in some cases, most likely as the result of serial mutations within the dominant B-cell clone. This implies that patients with persistent CLH require regular monitoring for the development of lymphoma.22 Clonal T-cell receptor gene rearrangements have also been detected in up to one-third of CLH cases.28,33 This may represent an expanded reactive T-cell clone within the CLH infiltrate. Evidence for this has been observed in CLH related to human immunodeficiency virus infection.34 Other possibilities include a low-grade T-cell lymphoma such as the CD4+ small/medium pleomorphic type17,35 or an evolving B-cell lymphoma bearing T-cell receptor gene rearrangements.
The differential diagnosis of CLH includes all of the conditions listed in Box 146-1. Many of these entities, such as granuloma faciale, Merkel cell tumor, histiocytomas, granulomas, and metastatic carcinoma, are readily distinguished from CLH by their histopathologic features.
Most Likely
Consider
Always Rule Out
|
A principal challenge in the differential diagnosis of CLH is its distinction from CBCL of either the diffuse or follicular types (see Chapter 145).4–6,27,36 The most common variant of CBCL is diffuse large cell lymphoma, which is readily distinguished from follicular CLH on the basis of its diffuse architecture and monomorphous large cell composition. Differentiating follicular CLH from follicular CBCL can be difficult. Follicular CBCL tends to be more bottom heavy with adnexal infiltration and lymphoid follicles lacking the cellular heterogeneity, zonation, and macrophages typical of reactive follicles. Immunophenotyping shows that neoplastic B-cell follicles are either monotypic or immunoglobulin negative.18–20 Cases involving cutaneous lymphoid infiltrates that are hard to classify or that contain a dominant lymphoid clone often benefit from a staging workup as described in Section “Approach to the Patient.” In those instances representing lymphoma, workup will sometimes reveal extracutaneous involvement, and biopsy specimens from these lesions may be easier to diagnose as lymphoma. Staging also provides useful prognostic data and is essential for appropriate management of patients with overt lymphoma.
CLH must also be distinguished from other disorders with prominent lymphocytic infiltrates such as chronic lymphocytic leukemia, deep figurate erythemas, polymorphous light eruption, discoid lupus erythematosus, and Jessner’s lymphocytic infiltration of the skin. All of these disorders tend to produce perivascular, and sometimes periadnexal, infiltrates whose density is far lower than that of the nodular or diffuse infiltrates typical of CLH. With the exception of chronic lymphocytic leukemia, these are all T-cell disorders. Chronic lymphocytic leukemia is a B-cell disorder that exhibits monotypic immunoglobulin and CD5 expression and involves the blood and bone marrow. CLH lesions rich in plasma cells must be distinguished from the lesions of secondary syphilis, plasmacytoma, myeloma, and immunocytoma. The infiltrate in secondary syphilis is generally richer in plasma cells or histiocytes than in small lymphocytes and is associated with epidermal hyperplasia, an interface inflammatory component, and positive results on serologic tests. The plasma cells in plasmacytoma and myeloma are monotypic and tend to exhibit mitotic figures, atypia, immaturity, and multinucleated forms. Monotypic immunoglobulin is also present in the tumor cells of primary cutaneous immunocytoma. This B-cell neoplasm is also known as marginal zone lymphoma, mucosa-associated lymphoid tissue (MALT)-type lymphoma, and low-grade skin-associated lymphoid tissue (SALT) B-cell lymphoma.5,27,37 It is composed of varying proportions of small lymphocytes, plasmacytoid lymphocytes, plasma cells, and marginal zone-like B cells. The latter have monocytoid features with a halo of cytoplasm visible around their nuclei. These cells often surround clusters of small lymphocytes, imparting a “reverse reactive follicle” appearance with smaller cells centrally and the larger monocytoid cells peripherally.
Other disorders that can contain dense cutaneous infiltrates of both T cells and B cells include (1) lymphomatoid keratosis,38 (2) pseudolymphomatous folliculitis,39 (3) systemic immunoglobulin IgG4-related plasmacytic syndrome (SIPS),40,41 and (4) CD4+ small/medium pleomorphic T-cell lymphoma.17,35 The first three are readily distinguished from classic CLH on clinicopathological grounds. Although it can mimic CLH clinically and contain abundant B cells and plasma cells, CD4+ small/medium pleomorphic T-cell lymphoma is recognized by its predominance of CD4+ atypical lymphoid cells, frequent dominant T-cell clonality, paucity of reactive lymphoid follicles, and T-follicular helper cell phenotype.
It should be noted that typical reactive lymphoid follicles with germinal centers are not specific for CLH. They can be seen in some lymphomas (e.g., marginal zone lymphoma), Kimura disease, angiolymphoid hyperplasia with eosinophilia, Castleman disease, inflammatory pseudotumor of the skin, and morphea and as part of the host response to tumors such as basal cell carcinoma. The broad histopathologic differential diagnosis of CLH also includes histiocytoses, especially Rosai–Dorfman disease (sinus histiocytosis with massive lymphadenopathy; see Chapter 148), as well as so-called small round cell tumors such as Merkel cell carcinoma (see Chapter 120), oat cell carcinoma, neuroblastoma, and Ewing sarcoma.
Differentiation between T-cell CLH and T-cell lymphomas relies on the fact that most cases of mycosis fungoides exhibit marked epidermotropism, and most nonmycosis fungoides cutaneous T-cell lymphomas are diffuse large cell types, although a pleomorphic small/medium cell type occurs rarely as discussed above. Many T-cell lymphomas show loss of one or more T-cell antigens, and most have monoclonal T-cell receptor gene rearrangements. Progression to T-cell lymphoma has been reported in occasional patients with T-cell CLH.19,20
One of the most challenging issues concerning cutaneous pseudolymphomas has been the nature of their relationship to true lymphomas. Many patients with CBCL have a history of prior diagnosis of biopsy specimens as CLH or atypical CLH. Patients with disorders such as lymphomatoid papulosis, lymphomatoid granulomatosis, and angioimmunoblastic lymphadenopathy with dysproteinemia have an increased risk of developing various forms of overt lymphoma.5,6,27,37 The advent of molecular biologic methods for determining B-cell or T-cell clonality by analyzing immunoglobulin or T-cell receptor gene rearrangements, respectively, has provided important insights into the relationship between pseudolymphomas and lymphomas.5,6,27 It appears that many forms of cutaneous pseudolymphoma represent the benign end of a lymphoproliferative continuum eventuating in true lymphoma at its malignant extreme. In between lies a spectrum of intermediate disorders that are often difficult to classify unless one conceptualizes the disorders along such a continuum. In some cases, such as the CLH–CBCL spectrum, lesions range from polyclonal CLH at the benign extreme, through an intermediate form known as clonal CLH, to overt CBCL at the malignant extreme. In other instances, such as the lymphomatoid papulosis–CD30+ lymphoma spectrum, dominant clonality is already present in most cases at the clinically benign end of the spectrum. The most compelling evidence for the validity of this concept of a continuum is that case studies verify that the same dominant lymphoid clone is involved in the clonal pseudolymphomatous precursor and the subsequent overt lymphoma when they arise in the same individual. This relationship between overt neoplasia and clonal precursor lesions is not unique to cutaneous pseudolymphomas. It is also seen in the connection between other forms of extranodal lymphoid hyperplasia and overt lymphoma and in the relationship between monoclonal gammopathy of uncertain significance (formerly termed benign monoclonal gammopathy) and subsequent B-cell malignancies.
The concept of a spectrum for cutaneous pseudolymphomas and associated lymphomas has several important implications for the diagnosis, management, and pathogenesis of these disorders. First, because some cases will exhibit lesions at an intermediate point along the lymphoproliferative continuum, they may be difficult to classify neatly into a specific disease entity. It is probably more important to recognize that they belong to a particular disease spectrum, because it is the spectrum that is often the most relevant factor for determining optimal therapy as well as the magnitude and nature of the subsequent lymphoma risk. Second, because many patients with cutaneous pseudolymphomas are at increased risk for a clonally related lymphoma, they should have regular clinical follow-up, and all reasonable attempts should be made to eradicate or suppress their disease. Third, patients with pseudolymphomas probably have underlying abnormalities in the regulation of lymphoid proliferation and/or clearance (e.g., via apoptosis). Clonal evolution and concomitant clinical disease progression are likely to occur through a sequence of somatic mutations that act through these mechanisms to confer progressively increasing autonomy to the dominant lymphoid clone. Elucidating these pathogenetic mechanisms will be important not only for developing novel therapies for cutaneous pseudolymphomas but also for improving the treatment of analogous lymphoproliferative disorders occurring in other organ systems.
CLH lesions may resolve spontaneously or persist indefinitely. Nodular scabies (see Figs. 146-3 and 146-6) is a well-recognized form of persistent CLH. Solitary lesions sometimes regress after biopsy or may be eradicated by excision. Those lesions that are secondary to drug use usually regress when the offending agent is withdrawn.
CLH related to infection with Borrelia burgdorferi (lymphocytoma cutis; see Chapter 187) responds to antibiotic therapy with cephalosporins, as do some cases of idiopathic CLH. Glucocorticoids (topical, intralesional, systemic), cryotherapy, antimalarials, minocycline, and radiation therapy have all been used with variable success (Box 146-2). The latter is particularly effective but is usually regarded as a treatment of last resort. Laser therapy and photodynamic therapy have also been beneficial in some cases.43,44 Local or distant recurrence can arise after any of these treatments. Cases of atypical CLH that cannot be distinguished from lymphoma are usually treated as localized lymphoma after staging rules out extracutaneous disease. Treatment typically involves local radiation therapy, which may be preceded by excision of the lesion.
FIRST LINE
SECOND LINE
|
Kimura Disease and Angiolymphoid Hyperplasia with Eosinophilia
The skin lesions of Kimura disease and angiolymphoid hyperplasia with eosinophilia (AHLE) most commonly affect young to middle-aged adults. Kimura disease is more common in Asian men, whereas AHLE is more common in women.50–53
It is possible that Kimura disease represents a florid, subcutaneously deep-seated form of the same basic pathogenetic process that gives rise to classic dermal CLH. Most favor the concept that Kimura disease and AHLE are distinct clinicopathologic entities despite some clinicopathologic overlap.54–56 The terms epithelioid hemangioma and pseudopyogenic granuloma have been used as synonyms for AHLE. Some regard AHLE essentially as a malformation of blood vessels caused by an underlying arteriovenous shunt. They consider the CLH-like aspects of the lesional infiltrate to be a secondary feature. There is also evidence of lymphatic vessel proliferation in AHLE.52 Compared with the lesions of Kimura disease, AHLE lesions tend to be smaller (Fig. 146-11
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


