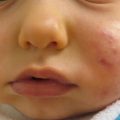
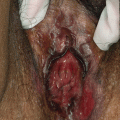
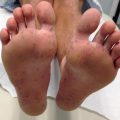
Fig. 23.1
Alternating bands of light and dark color along hair fibers indicative of periods of protein deficiency in phenylketonuria (Courtesy of Dr. Maureen Rogers)
Specific Investigations
Newborn screening (tandem mass spectrometry)
Plasma amino acid analysis
Molecular genetic testing
PKU can be diagnosed by newborn screening in virtually 100 % of cases using tandem mass spectrometry. Elevated Phe concentrations in blood spots can be quantified as early as 24 h after birth, and tyrosine (Tyr) concentrations can be used to calculate a Phe:Tyr ratio. Plasma amino acid analysis is the standard method for confirming elevated Phe in newborns having a positive newborn screen. PKU is diagnosed in individuals with plasma Phe concentrations higher than 1,000 μmol/L in the untreated state. Molecular genetic testing of PAH is used primarily for genetic counseling purposes to determine carrier status of at-risk relatives and for prenatal testing.
Table 23.1
First-line therapies
Diet (Phe-restricted diet) | B (level of evidence) |
A Phe-restricted diet and a Phe-free medical formula should be started as soon as possible after birth under the direction of a nutritionist. Intake of tyrosine and total amino acids must be monitored. Children under age 2 years should maintain a total amino acid intake of at least 3 g/kg/day including 25 mg tyrosine/kg/day. Care must be taken to avoid long periods of low blood Phe concentration, which is also harmful to brain development.
Reduction of phenylalanine levels by dietary restriction can prevent neurocognitive dysfunction, although a mild reduction in IQ (when compared to siblings) and defect in executive function can be present even in optimally treated subjects [1].
Table 23.2
Second-line therapies
Sapropterin dihydrochloride | A (level of evidence) |
Large neutral amino acids (LNAA) | A (level of evidence) |
Although the treatment of PKU with phenylalanine-restricted diets has been hugely successful, the poor palatability of the diet results in poor compliance in adolescence and adulthood. A number of attempts to find other treatment modalities for PKU are ongoing. Patients with the milder variants are more likely to respond to pharmacological treatment with sapropterin, a synthetic analog of tetrahydrobiopterin [2].
Acrodermatitis Enteropathica
Acrodermatitis enteropathica (AE) is an autosomal recessive disorder caused by a mutation in the SLC39A4 gene, resulting in reduced synthesis of the intestinal zinc transporter, ZIP4. This results in defective absorption of dietary zinc, leading to severe zinc deficiency. AE occurs worldwide, with an estimated incidence of 1 per 500,000 children, and usually presents within days in bottle-fed infants and days to weeks after weaning in breast-fed infants. AE presents with erythematous eczematous scaly plaques over the extremities, anogenital and periorificial skin and can become vesicular, bullous, pustular, desquamative, and erosive (Figs. 23.2 and 23.3). Without treatment, patients can develop generalized alopecia and diarrhea. In later stages, clinical features include growth and developmental delay, poor wound healing, anemia, photophobia, hypogeusia, anorexia, secondary infections, delayed puberty, and hypogonadism in males. Acquired zinc deficiency secondary to insufficient intake, excessive losses, or malabsoption can present with identical clinical features.
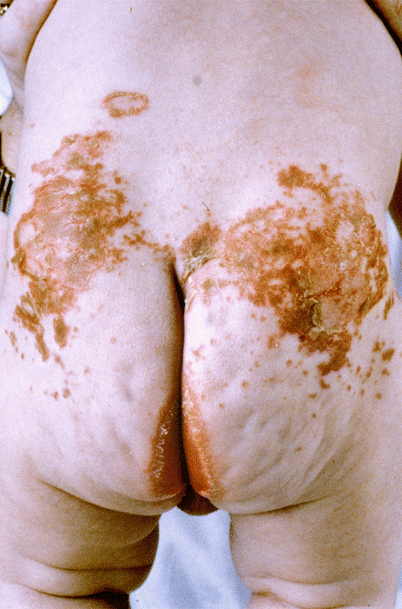
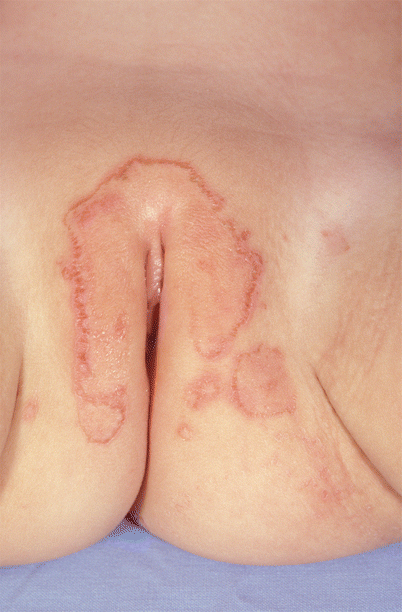
Fig. 23.2
Erythematous erosive scaly plaques over the lower back and perianal skin in a patient with acrodermatitis enteropathica (Courtesy of Dr. Maureen Rogers)
Fig. 23.3
Erythematous erosive scaly plaques over the anogenital skin in a patient with acrodermatitis enteropathica (Courtesy of Dr. Maureen Rogers)
Specific Investigations
Plasma zinc concentration
Alkaline phosphatase
Histopathology
DNA analysis
The diagnosis of AE is made through recognition of the clinical presentation, supported by histopathology and laboratory tests. Zinc levels can be determined by measurement of plasma or serum zinc concentrations, which is ideally drawn before breakfast, in a trace element-free collection tube. Pre-breakfast zinc levels should be greater than 70 ug/dL. Subsequent meals during the day will lower this value. A serum zinc level of less than 50 ug/dL is suggestive of AE [3]. A low serum alkaline phosphatase, a zinc-dependent enzyme, can support the diagnosis of zinc deficiency. The histopathologic features of AE classically show “necrolysis,” a term describing cytoplasmic pallor, vacuolization, ballooning degeneration, and confluent necrosis of keratinocytes within the superficial stratum spinosum and stratum granulosum of the epidermis. In resolving or chronic AE lesions, psoriasiform hyperplasia is present, and necrolysis may be minimal to absent.
Table 23.3
First-line therapies
Zinc supplementation | C (level of evidence) |
Zinc replacement therapy should be started at 3 mg/kg/day of elemental zinc (there is 50 mg of elemental zinc per 220 mg of zinc sulfate). Typically, clinical improvement is seen very rapidly, within days to weeks, even before a significant change in serum zinc levels. Serum or plasma zinc levels and zinc-dependent enzyme levels should be monitored every 3–6 months and the dose of zinc sulfate should be adjusted appropriately. Acute zinc overdose may cause diarrhea, light-headedness, gait disturbances, lethargy, vomiting, epigastric pain, and abdominal cramps. Chronic overdose may lead to neutropenia, leukopenia, copper and/or iron deficiency, anemia, growth retardation, decreased high-density lipoprotein, and increased low-density lipoprotein.
Biotin Deficiency
Biotin deficiency may be acquired or inherited. The inherited form of biotin deficiency is due to innate errors of biotin metabolism caused by a deficiency in holocarboxylase synthetase (HCS), biotinidase (BTD), or the sodium-dependent multivitamin transporter (SMVT). All three lead to multiple carboxylase deficiency and manifest with metabolic acidosis, neurological abnormalities, and a skin eruption. Acquired biotin deficiency occurs as a result of inadequate intake or excessive losses.
Patients with inherited biotin deficiency typically present within the first weeks to first year of life. The most common symptoms are developmental delay, hypotonia and seizures. The cutaneous findings usually consist of an erythematous and scaly eruption typically localized to moist and periorificial areas. Other skin findings include alopecia and sparse hair, cutaneous grayish pallor and atrophy of the lingual papillae. Other symptoms include conjunctivitis, blepharitis, optic atrophy, metabolic acidosis, vomiting, lethargy, ataxia, and hearing loss.
Biotinidase deficiency should be considered in children with skin eruption or hair loss who also show neurologic dysfunction, especially infantile seizures, or unexplained breathing problems associated with keto-lactic acidosis, organic aciduria, and hyperammonemia.
Failure to diagnose and treat BTD deficiency at an early stage may cause irreversible neurological damage, leading to developmental delay and autistic behavior.
Specific Investigations
Serum and urinary biotin levels
Biotinidase, holocarboxylase synthetase and sodium-dependent multivitamin transporter activity
Laboratory evaluation will show decreased biotin levels. Biotinidase deficiency can be diagnosed by demonstrating decreased or absent enzyme activity in serum, peripheral blood leukocytes, or cultured skin fibroblasts. Prenatal diagnosis is also possible because biotinidase activity can be measured in cultured amniotic fluid cells.
Table 23.4
First-line therapies
Oral biotin | C (level of evidence) |
In cases of biotin deficiency, treatment includes oral biotin supplementation with doses of 5–40 mg daily [4]. Skin and neurologic symptoms improve rapidly if biotin therapy is initiated early. However, if treatment is delayed, neurologic dysfunction may be permanent. In inherited forms of biotin deficiency, biotin therapy must be continued throughout life.
Organic Acidemia
Organic acidemias are a diverse set of metabolic conditions which include maple syrup urine disease (MSUD), methylmalonic acidemia (MMA), and propionic acidemia. These congenital branched-chain amino acid metabolism disorders lead to protein intolerance and can unfrequently cause a generalized exfoliative erythroderma or a periorificial and acral eruption similar to acrodermatitis enteropathica [5].
MSUD is caused by a deficiency of the branched-chain keto acid dehydrogenase complex and results in elevated leucine, isoleucine, and valine concentrations in blood, urine, and tissues. Poor feeding, vomiting, respiratory problems, lethargy, hypotonia, and seizures are the main features of MSUD that appear during the neonatal period. Methylmalonic acidemia is caused by a reduction in the activity of methylmalonic coenzyme A (CoA) mutase or its coenzyme adenosylcobalamin. Propionic acidemia is caused by a deficiency of propionyl CoA carboxylase or abnormal metabolism of its coenzyme, biotin. The clinical features of both MMA and propionic acidemia are similar; poor feeding, hypotonia, vomiting, metabolic acidosis, and lethargy, which may result in neonatal death. Cutaneous lesions are uncommon, but can include superficial desquamation, periorificial dermatitis, psoriasiform eruptions, and alopecia.
Specific Investigations
Urine organic acids
Specific enzyme activity assay
Molecular testing
Laboratory findings that point to an organic acidemia include acidosis, ketosis, hyperammonemia, and abnormalities in liver synthetic function. Organic acidemias can be identified though measurement of urine organic acids using gas chromatography with mass spectrometry. When done in times of illness, the diagnostic yield is higher. Confirmatory testing can be done through assay of the activity of the deficient enzyme in lymphocytes or cultured fibroblasts and/or molecular genetic testing.
Table 23.5
First-line therapy
Low protein diet | C (level of evidence) |
Table 23.6
Second-line therapy
Oral thiamine (MSUD) | E (level of evidence) |
Hydroxocobalamin (MMA) | E (level of evidence) |
Oral antibiotics (propionic acidemia) | E (level of evidence) |
Table 23.7
Third-line therapy
Liver and kidney transplantation | E (level of evidence) |
In the organic acidemias, a low-protein diet with supplementation of essential amino acids is the therapy of choice [6]. The use of specific formulas deficient in the particular precursor amino acids for each disorder is a critical part of management, as it provides the essential amino acids in an otherwise protein-deficient diet. However, if the diet is strictly limited in branched-chain amino acids, the most critical one being isoleucine, cutaneous lesions resembling acrodermatitis enteropathica may also result. In select cases, adjunctive compounds can be used to reduce the burden of toxic metabolites and/or increase activity of deficient enzymes. Examples would be oral thiamine in select cases of MSUD, hydroxocobalamin in MMA, and antibiotics to decrease the production of propionate by gut bacteria in propionic acidemia. Liver and kidney transplantation is uncommonly used to treat cases of MMA and propionic acidemia.
Kwashiorkor
Also known as “protein energy malnutrition,” kwashiorkor is a prevalent disease with a high mortality rate of 30–50 %. Approximately 30,000 children die each day from kwashiorkor, mostly children ages 0–4 who live in Africa. Death is usually secondary to electrolyte imbalance from uncontrollable diarrhea or infection. Kwashiorkor is defined by 60–80 % predicted body weight for age and height, plus edema and/or hypoalbuminemia. Multiple social and economic factors influence its prevalence and distribution, including poverty and resource allocation in third-world countries, and fad diets, food avoidance (for perceived allergies), and food withdrawal (child abuse) in the United States. The disease onset in developing countries coincides with the discontinuation of breastfeeding.
Social chaos and disruptive family dynamics are common in the household of kwashiorkor patients, regardless of the underlying cause of malnutrition. Patients with protein malnutrition who appear to live in a stable home environment with adequate, consistent food sources should be investigated for medical causes for protein malabsorption of the gastrointestinal tract, as cystic fibrosis and Crohn’s disease have been documented causes of kwashiorkor.
While insufficient protein intake is the presumed primary etiology of this condition, many other factors may play a role. Several hypotheses support this notion, especially since total caloric deprivation (marasmus) has a lower mortality rate than kwashiorkor. Aflatoxins in food, poor innate protein synthesis, and increased susceptibility to infection are suggested perpetuating factors. The “free radical theory” proposes these processes saturate the serum and tissue with free radicals, depleting the patient’s internal antioxidant stores. This theory is supported by increased serum markers of oxidative stress and low antioxidant levels during active disease, and normalization of levels after treatment.
Physical examination will show an apathetic, irritable child with profound delay in developmental milestones. The patient will oftentimes have an overall pallor to the skin, remarkably lighter than other family members. A prominent exam finding is red-brown patches and plaques of inflamed skin with a hyperkeratotic rim, called “flaking paint dermatitis.” This dermatitis is most prominent along the trunk, and spares the dorsal hands and feet. Alternating bands of scalp hair pigment, the “flag sign,” may be present. Loss of hair pigment signifies times of greater protein restriction.
Specific Investigations
Serum chemistries for protein deficiency (albumin)
Serum chemistries for anemia (CBC, iron level, TIBC)
Serum chemistries for electrolyte imbalance (Na, K, Ca, Cl)Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree