Chapter 28 Hypophosphatemia
 Access the complete reference list online at http://www.expertconsult.com
Access the complete reference list online at http://www.expertconsult.com
 IN THIS CHAPTER
IN THIS CHAPTER  PowerPoint Presentation Online
PowerPoint Presentation Online
Introduction
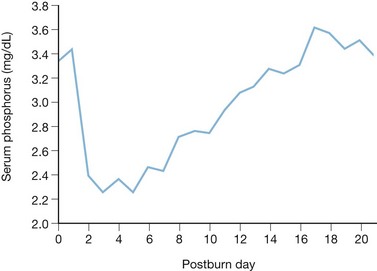
Certain humoral and metabolic responses to thermal and mechanical trauma that maintain homeostasis and prevent cellular dysfunction also produce alterations in electrolyte balance. An example is renal retention of sodium during the resuscitative phase of burn injury, which alters sodium balance in the course of preserving intravascular volume. Despite the markedly increased cardiac output and renal plasma flow that occur in the subsequent flow phase, a decrease in blood volume persists and results in sustained elevation of plasma renin activity, secretion of antidiuretic hormone, and sodium retention.1 Conversely, the severe hypophosphatemia that often follows major injury occurs concomitantly with a 50–100% increase in resting energy expenditure, leading to a possible deficiency in the high-energy phosphate compounds essential for cellular metabolism. Thermal injury induces a precipitous decrease in serum phosphate concentration that reaches its nadir between the second and fifth post-burn days. This phenomenon has been recognized for quite some time2 and was recently confirmed by the authors in a large series of burn patients.3 Despite aggressive phosphorus supplementation, normal levels of serum phosphorus are rarely reached prior to the tenth post-burn day (Fig. 28.1). Of 550 patients studied, 175 had serum phosphorus concentrations below 2.0 mg/dL, and of these 49 were below 1.0 mg/dL, with the lower limit of normal serum phosphorus being 3.0 mg/dL. Such hypophosphatemia is not exclusive to thermal injury, having been described following multiple trauma,4 head injury,5 and elective surgery.6 The exact mechanism by which thermal injury or severe stress induces hypophosphatemia is unknown. Several events associated with burn injury, however, affect phosphorus metabolism, and these may combine to produce hypophosphatemia.
Etiology of post-burn hypophosphatemia
Many of the pathophysiological changes and therapeutic interventions that occur during the first post-burn week influence serum phosphorus concentration (Box 28.1). Hypophosphatemia does not necessarily imply phosphorus depletion; in the case of burn injury, most patients are healthy prior to injury and presumably have normal phosphorus stores. Nor do simple calculations of phosphate balance explain the dramatic decrease in serum levels; simultaneous reduction of urinary phosphate excretion is observed, suggesting an extrarenal mechanism. The fractional excretion of phosphate, however, increases during the early period of diuresis following burn injury (post-burn days 2–4), potentially contributing to the decline in serum levels. The pathophysiological events and therapeutic interventions discussed below are associated with hypophosphatemia in other disease states and in certain experimental animal models, but the extent of their contributions to the post-burn decrease in serum phosphorus has not been critically evaluated and is, at present, undefined.
Stress response
In the early post-burn period, the classic ‘fight or flight’ response occurs, with elevation of plasma catecholamines, glucose, glucagon, and cortisol. Exogenous epinephrine administration has been associated with the development of hypophosphatemia, and the profound catecholamine release accompanying thermal injury may contribute to the early decrease in serum phosphorus. The mechanism by which this occurs is uncertain but may be a consequence of the accompanying hyperglycemia, resulting in a redistribution of phosphorus from the extracellular to the intracellular compartment (see section on Metabolic support, below). In acute clinical states of glucagon excess, tubular reabsorption of phosphate is impaired in both the proximal and distal nephron, leading one to expect renal phosphate wastage.7 Since urinary excretion of phosphate is usually decreased in the early post-injury period, the importance of hyperglucagonemia remains uncertain. Administration of pharmacological doses of glucocorticoids enhances phosphorus excretion and impairs phosphate absorption by the gut and reabsorption by the kidney. Whether or not the adrenocortical response significantly contributes to the hypophosphatemia after burn injury is not known.
Resuscitation and topical therapy
Administration of large doses of sodium lactate for initial burn resuscitation may decrease the serum phosphorus concentration by several mechanisms.8 Lactate is converted to glucose in the liver, a process requiring high-energy phosphate availability. Additionally, though it does not usually occur clinically, metabolic alkalosis induced by lactate infusion may result in depression of serum phosphorus concentration. Alkalosis is associated with an increase in glycolysis that promotes transfer of phosphorus to the intracellular space. During resuscitation, alkalemia is uncommon and patients are more likely to manifest a mild metabolic acidosis, which is compensated by hyperventilation, resulting in a normal or mildly alkaline blood pH. Acidosis markedly inhibits renal phosphate reabsorption, resulting in phosphaturia. The contribution of this mechanism to post-burn hypophosphatemia is probably minor; early renal phosphate wastage is not observed, perhaps being obscured by diminished glomerular filtration early in burn injury. In addition, the p-carboxy metabolite of mafenide acetate strongly inhibits carbonic anhydrase. Such inhibition diminishes proximal tubular reabsorption of phosphate and probably occurs following topical burn wound treatment with mafenide, but the magnitude of the effect is unknown.
Expansion of the extracellular fluid volume is also associated with inhibition of proximal tubular phosphate reabsorption. A tight coupling exists between sodium and phosphate transport across the renal epithelial cell. In patients with burns, mobilization and excretion of the large edema volume usually begins by the second post-burn day and continues throughout the next week to 10 days. In contrast to the relative paucity of phosphate excretion during the first 24 h after injury, when glomerular filtration is markedly reduced, a modest loss of phosphate may occur with diuresis of the edema fluid. In fact, the diuretic phase is associated with an increase in the fractional excretion of phosphate despite a concomitant reduction in the serum phosphate concentration.9 Phosphorus excretion during the natriuretic phase of early burn injury is consistent with the tight coupling observed in other diuretic states.
Ulcer prophylaxis
Effective prophylaxis against Curling’s ulcers with H2-antagonists and antacid buffering has been a mainstay of burn care for the past two decades. Significant degrees of hypophosphatemia and phosphate depletion occur during continuous or chronic administration of phosphate-binding agents containing magnesium, calcium, and aluminum. These agents bind not only dietary phosphate but also phosphate secreted into the intestinal lumen, often resulting in a net negative phosphate balance. The severity of such hypophosphatemia clearly depends on the dose of phosphate-binding agents, dietary phosphorus intake, and pre-existing phosphate balance. To reduce alimentary scavenging of dietary and secreted phosphate, buffering with antacids containing aluminum phosphate salts (Al2PO4), which do not bind any additional phosphate, may be utilized. Sucralfate, which is also effective in preventing upper gastrointestinal stress ulceration following thermal injury, is not a buffering agent, but as a complex salt of aluminum hydroxide, is capable of binding phosphate. Its administration has also been associated with the development of hypophosphatemia in critically ill patients.10
Hyperventilation
Respiratory alkalosis is often present during the first week post-burn and may be enhanced by anxiety or pain, and even by the inhibition of carbonic anhydrase induced by mafenide acetate burn cream. As fluid resuscitation progresses, respiratory rate and tidal volume progressively increase, resulting in minute ventilation that may be twice normal. Mild hyperventilation induces only a slight decline of serum phosphorus levels; prolonged, intense hyperventilation, however, may result in serum phosphorus values less than 1.0 mg/dL.11 During respiratory alkalosis, phosphorus virtually disappears from the urine, eliminating renal losses as the causative mechanism. Respiratory alkalosis induces a rapid movement of carbon dioxide from the intracellular to the extracellular space. Intracellular pH increases, activating glycolysis and increasing the formation of intracellular phosphorylated carbohydrate compounds. The readily diffusible inorganic phosphate pool supplies the required phosphorus, and serum phosphorus concentrations consequently fall abruptly. The extent to which this mechanism contributes to post-burn hypophosphatemia is uncertain.
Metabolic support
Administration of carbohydrates may play a major role in the development of post-burn hypophosphatemia. Infusion of glucose solutions or oral intake of carbohydrates produces mild hypophosphatemia in healthy individuals. This decrease in serum phosphate is associated with an increase of inorganic phosphate, ATP, and glucose 6-phosphate in muscle cells. The mechanism by which such carbohydrate administration induces hypophosphatemia is somewhat speculative. Experience with phosphate-deficient total parenteral nutrition and subsequent development of hypophosphatemia has provided some insight into the etiology.12,13 As carbohydrates are absorbed, insulin secretion increases, shifting phosphorus from the extracellular to the intracellular space. If phosphate reserve is low, ATP is poorly regenerated since hypophosphatemia inhibits glucose 3-phosphate dehydrogenase. Inorganic phosphates in the intracellular pool become further diminished because of incorporation, initially as newly synthesized ATP, but eventually as triose phosphates when the ATP is consumed in the hexokinase reaction. Glucose utilization by red blood cells requires ATP at the hexokinase and phosphofructokinase steps, but regeneration of ATP does not occur during phosphate deficiency or acute hypophosphatemia due to a block at the glucose 3-phosphate dehydrogenase step. In states of phosphate depletion, the scant phosphate that enters the red blood cell is incorporated into 1,3-diphosphoglycerate, but most is diverted to 2,3-diphosphoglycerate, also preventing complete glycolysis to regain the ATP consumed.
In thermally injured patients, infusion of dextrose-containing solutions usually begins 24 h post-burn, and enteral nutrition, in which most of the calories are supplied as carbohydrates, is initiated within several days of injury. These interventions are temporally correlated with the rapid descent of serum phosphorus concentrations. In other clinical states, severe hypophosphatemia following the initiation of enteral or parenteral nutrition is most commonly associated with the feeding of patients with advanced protein-calorie malnutrition. When total body phosphorus is depleted by starvation, serum phosphorus levels usually remain normal, but carbohydrate administration produces a rapid marked decline in serum phosphorus concentration. If untreated, this may result in multi-system organ dysfunction, respiratory and cardiac failure, or death. Thermally injured patients are usually well nourished prior to burn injury and the clinical scenario of refeeding hypophosphatemia may not apply to them. Similar findings, however, have been described recently in previously well-nourished surgical intensive care unit patients in whom the initiation of isotonic enteral feedings resulted in a decrease of serum phosphorus from normal levels to approximately 1 mg/dL, a level that is considered to be dangerously low and to require prompt supplementation.14,15
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


