Histiocytic Neoplasms
Michael A. Cardis
A. Yasmine Kirkorian
Alejandro A. Gru
INTRODUCTION
A recent update in the classification of histiocytic disorders of adults and children has been proposed, which provides integration of cell of origin of these disorders, in addition to common molecular pathways that appear to be altered in this group of neoplasms. To this extent, histiocytoses can be divided into five major categories: (1) Langerhans related (“L”); (2) cutaneous and mucocutaneous (“C”); (3) malignant histiocytoses (“M”); (4) Rosai-Dorfman disease (RDD; “R”); and (5) hemophagocytic lymphohistiocytosis and macrophage activation syndrome (“H”).1 The “L” group includes Langerhans cell histiocytosis (LCH), indeterminate cell histiocytosis (ICH), Erdheim-Chester disease (ECD), mixed LCH/ECD, and the disseminated forms of juvenile xanthogranuloma (JXG). They all share clonal mutations involving genes in the MAPK pathway in >80% of cases; the “C” group incorporates the cutaneous xanthogranulomas (JXG, adult xanthogranuloma, solitary reticulohistiocytoma [SRH], benign cephalic histiocytosis [BCH], generalized eruptive histiocytosis [GEH], and progressive nodular histiocytosis [PNH]) and non-xanthogranulomatous disorders (cutaneous RDD and necrobiotic xanthogranuloma). The “R” group includes familial RDD, in addition to the sporadic extracutaneous forms (classical, extranodal, etc). The “M” group is an extraordinary rare group of disorders that include histiocytic, interdigitating dendritic cell, Langerhans cell, or indeterminate cell sarcomas. Finally, the “H” group includes the primary and secondary hemophagocytic lymphohistiocytosis. In this chapter, we will only focus on the diseases that primarily affect the skin.
GROUP “L” HISTIOCYTOSES
LANGERHANS CELL HISTIOCYTOSIS
Definition and Epidemiology
LCH (formerly histiocytosis X) represents a clonal proliferation of immature dendritic cells expressing CD1a and CD207 (langerin—surrogate marker for the presence of Birbeck granules) producing a clinical picture ranging from a focal lesion to disseminated disease affecting numerous organ tissues.2 LCH was initially described as three different diseases, each with a unique clinical picture: eosinophilic granuloma, Hand-Schüller-Christian disease, and Letterer-Siwe disease. Although the aforementioned nomenclature has been abandoned for the use of LCH or Langerhans cell disease, they are still useful to illustrate the various clinical courses that LCH can encompass.3,4
Langerhans cell proliferations include reactive Langerhans cell hyperplasia, LCH, Langerhans cell sarcoma (LCS), and congenital self-healing reticulohistiocytosis (CSHR).5,6 Debate still lingers on the nature of LCH, almost 20 years after the publication of the study with the demonstration of clonality in nonpulmonary LCH lesions.7 Newer data now show evidence of a clonal myeloid cell origin for LCH.8,9,10,11,12,13,14 Despite a rare overall incidence in the general population, LCH has a strong predilection for children and particularly an aggressive clinical course in the very young individuals, typically less than 2 years of age at diagnosis.7,15,16 Nowadays, LCH is considered the prototypic histiocytosis from the “L” group.
LCH is most common in children and in white individuals of northern European ancestry and is usually diagnosed before age 4.3,17 The estimated incidence of LCH in patients less than 15 years old is 4 to 5 cases per million per year.3,18 LCH has a slight male predominance and is considered a sporadic disease, although a familial predisposition has been reported.19
Etiology
There has been much debate as to whether LCH represents a reactive or neoplastic illness. Evidence to support the former includes the following: LCH lesions may spontaneously resolve, there is no evidence as to the immortalization of the LCH cells in the laboratory, and proinflammatory cytokines and chemokines contribute to the pathogenesis of LCH.3 However, in 2010, it was discovered that in a large proportion of LCH cases (greater than half of cases), there is a gain-of-function somatic mutation in lesional cells involving BRAF (V600E), resulting in uncontrolled activation of the MAPK pathway (RAS/RAF/MEK/ERK).20 It was also noted that even in wild-type BRAF cases of LCH, ERK remained phosphorylated, suggesting that even in those cases other mutations (such as MAP2K1) in the same pathway are responsible for the disease. Indeed, MAPK is active in all LCH lesions, and LCH involves a clonal proliferation of cells, thus providing strong evidence that LCH is a neoplastic disease. Berres et al found a prevalence of 64% for BRAF mutations in LCH and particularly a high risk of recurrence among the mutated cases.11 Héritier et al found that the mutation showed correlation with high-risk features, increased risk of recurrence, and increased resistance to first-line therapies.21 Whole-exome sequencing subsequently identified the presence of MAP2K1 (MEK1) mutations in 7 of 21 cases of LCH with wild-type BRAF and in none of 20 BRAF V600E mutant cases. These studies implicate aberrations of the MAPK signaling pathway in the pathogenesis of LCH and strongly support a neoplastic pathogenesis.22,23,24,25
It has also recently been proposed that the timing and/or location at which point the disease-initiating cell acquires the mutation dictates the extent of disease, that is, mutations in premature CD34+ progenitor cells of the hematopoietic system lead to severe disease, whereas mutations in more differentiated cell such as lesional CD207+ cells in the skin lead to more localized disease.2,26 The propensity for LCH to induce osteolytic lesions is thought to result from an interaction between T-lymphocytes and lesional LCH cells, causing production of cytokines that stimulate the differentiation and activation of osteoclasts that produce destructive enzymes.
Clinical Presentation
The clinical presentation of LCH is broad and ranges from a solitary osteolytic lesion (eosinophilic granuloma/unifocal LCH) to multifocal bone disease that may include the triad of osteolytic bone lesions, diabetes insipidus, and exophthalmos (Hand-Schüller-Christian disease/multifocal unisystem LCH) to an aggressive, disseminated life-threatening form of disease (Letterer-Siwe disease/disseminated multifocal multisystem LCH). Those clinical presentations were later understood to represent a spectrum of the same illness that for a while was termed histiocytosis X, a term that was later replaced by LCH.26,27 The clinical classification of LCH is stratified into single organ system disease (SS-LCH) and multiorgan system disease (MS-LCH) in addition to unifocal versus multifocal involvement. Furthermore, MS-LCH is further classified according to whether or not there is involvement of risk organs such as the lung, liver, spleen, or bone marrow (RO-positive vs. RO-negative MS-LCH).3 SS-LCH represents up to 70% of cases, whereas only about 10% of cases end up with RO-positive MS-LCH.3 The most common site of involvement in SS-LCH is bone, in either a focal or multifocal pattern, followed by skin, lymph node, and lung.17,28 The bone is involved in about 80% of LCH patients and may either be asymptomatic or produce pain. In pediatric cases, the skull is the most commonly affected site, and on plain film shows punched-out osteolytic lesions with crisp margins. Organ system involvement in MS-LCH may include a wide array of systems, but prominent ones include the bone and skin followed by the liver, spleen, and bone marrow (hematopoietic system). Almost half of all LCH cases will involve the skin to some extent during the disease course, and up to 13% of patients will have disease exclusively involving the skin at the time of diagnosis.28 This variability in distribution and extent of involvement best explains the long list of clinical designations under the LCH umbrella.29
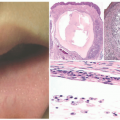
The most common presenting signs/symptoms of LCH are the following: soft tissue mass, bone pain, skin rash, fever, otorrhea, and/or lymphadenopathy.3 Skin findings in LCH are variable and can include numerous morphologies, among which the most notorious clinical picture is that of an erythematous, scaly dermatitis in a seborrheic distribution involving the scalp, postauricular skin, axillae, and diaper area. Other morphologic findings include “blueberry muffin”-like nodules, petechia, vesicles, or red to brown papules and plaques, often with secondary changes such as erosion, crusting, bleeding, or ulceration located in the scalp, ears, flexures, or acral sites.27,28,30 Ulcerations may occur in the axillary or inguinal folds. Isolated skin involvement by LCH is infrequent and occurs in <5% of all cases. Skin-limited LCH occurs in the form of one or multiple, often asymptomatic but occasionally erosive, papules or nodules with surrounding erythema or rarely forming masses with or without ulceration (Figure 26-1). One study documented mucosal involvement—most frequently the oral or genital mucosa—in 20% of cases with otherwise isolated skin LCH (Figure 26-2).31 The gingiva, when involved, presents with swelling or ulceration. Bone and central nervous system (CNS) involvement can produce serious sequelae such as exophthalmos, conductive hearing loss, loss
of teeth, and even spinal paralysis. If the hypothalamic-pituitary axis is involved, central diabetes insipidus may ensue, as may other endocrinologic or hypothalamic symptoms. Even with treatment and cure of disease, there can be permanent complications. Prognosis is based on involvement of high-risk organ systems (liver, spleen, lung, and bone marrow), as well as response to 6 weeks of standard treatment.2,32 Children with RO-positive MS-LCH have a mortality that ranges from 10% to 50%. Skin-limited disease portends an excellent prognosis.3
of teeth, and even spinal paralysis. If the hypothalamic-pituitary axis is involved, central diabetes insipidus may ensue, as may other endocrinologic or hypothalamic symptoms. Even with treatment and cure of disease, there can be permanent complications. Prognosis is based on involvement of high-risk organ systems (liver, spleen, lung, and bone marrow), as well as response to 6 weeks of standard treatment.2,32 Children with RO-positive MS-LCH have a mortality that ranges from 10% to 50%. Skin-limited disease portends an excellent prognosis.3
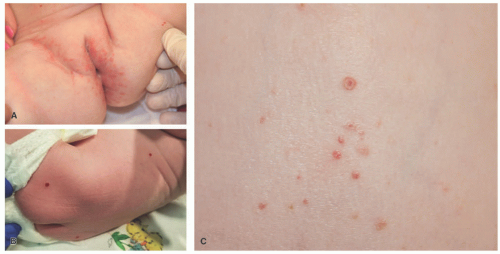 FIGURE 26-1. Langerhans cell histiocytosis (LCH). A, Multiple excoriated papules within the inguinal and vaginal area. B, Red-brown nodules in the back. C, The papules in LCH can have an umbilicated appearance. |
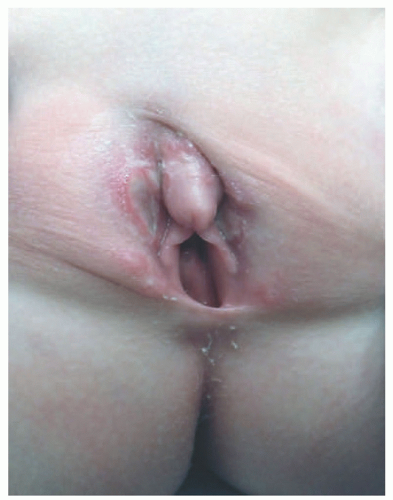 FIGURE 26-2. Vulvar erosions in Langerhans cell histiocytosis. |
Systemic symptoms including fever, lymphadenopathy, hepatosplenomegaly, and cytopenias can be seen in multisystem disease, together with clinical manifestations of organ dysfunction such as diabetes insipidus in cases with pituitary involvement. Such clinical manifestations warrant the staging of LCH using bone scans and magnetic resonance imaging of the brain, in addition to blood evaluation for electrolyte imbalances.
Histologic Findings
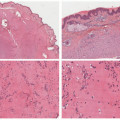
A common unifying theme in all of these lesions/clinical entities is the histologic demonstration of Langerhans cells in aggregates despite the admixture with eosinophils, macrophages, lymphocytes, neutrophils, and non-neoplastic multinucleated giant cells.33,34,35,36 The diagnosis of LCH can be tentatively made based on the characteristic morphologic findings of the Langerhans cells that are medium to large in size, exhibit abundant eosinophilic cytoplasm, and have a “coffee bean”-shaped grooved nuclei (Figures 26-3 and 26-4).37 They also have fine chromatin and indistinct, small nucleoli, and scattered mitoses may be visualized. These characteristic features although best seen on cytologic preparations can also be appreciated in a well-fixed and well-prepared histologic section. Involvement of the skin may be confined to small collections of cells at the dermal-epidermal interface with variable basal vacuolar degeneration or as dense infiltrates throughout the dermis with or without the formation of eosinophilic abscesses. Marked epidermotropism
is also frequently seen (Figure 26-5). Extravasated erythrocytes and a mixed inflammatory infiltrate with eosinophils, lymphocytes, neutrophils, plasma cells, and histiocytes (sometimes multinucleate giant cells, osteoclast like) are often present, and there are occasionally areas of focal necrosis.17 The LCH cells often aggregate into granulomas. As the time course of disease progresses, macrophages tend to predominate, and xanthomatous and fibrotic change may become prominent findings.
is also frequently seen (Figure 26-5). Extravasated erythrocytes and a mixed inflammatory infiltrate with eosinophils, lymphocytes, neutrophils, plasma cells, and histiocytes (sometimes multinucleate giant cells, osteoclast like) are often present, and there are occasionally areas of focal necrosis.17 The LCH cells often aggregate into granulomas. As the time course of disease progresses, macrophages tend to predominate, and xanthomatous and fibrotic change may become prominent findings.
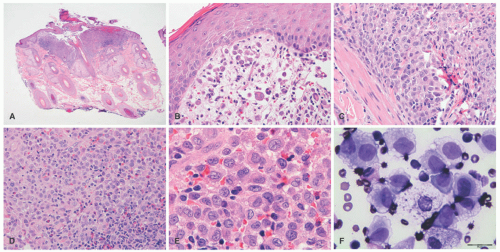 FIGURE 26-3. Langerhans cell histiocytosis (LCH)—histopathologic findings. A and B, The lesion has a folliculocentric pattern. C to E, Sheets of atypical Langerhans cells are seen with frequent nuclear grooves. F, Smear of LCH. |
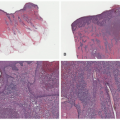
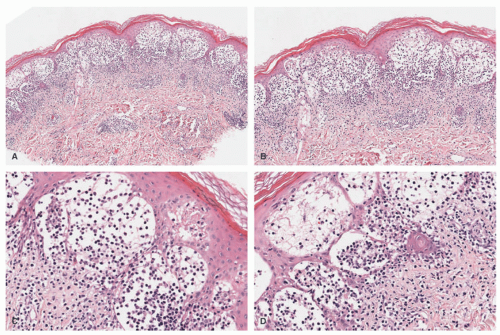 FIGURE 26-4. Langerhans cell histiocytosis—histopathologic findings. A and B, In this case, bullous formation is noted. C and D, Extensive epidermotropism is noted. Digital slides courtesy of Path Presenter.com. |
Confirmation of the diagnosis is achieved by demonstrating Langerhans granules (Birbeck granules) on electron microscopy38 or—more easily—by the characteristic positivity of Langerhans cells for CD1a, S-100, and langerin (Figure 26-5). The langerin immunostain is used, by many, as a surrogate marker for the presence of Birbeck granules.39,40,41,42,43 Langerin (CD207), a cell surface glycoprotein receptor of the lectin family indicative of Birbeck granule formation, is highly sensitive and specific for LCH. CD207 has mostly replaced ultrastructural studies (electron microscopy) in diagnosing LCH. Langerin has similar sensitivity, but slightly less specificity for LCH when compared to CD1a.3,33,44
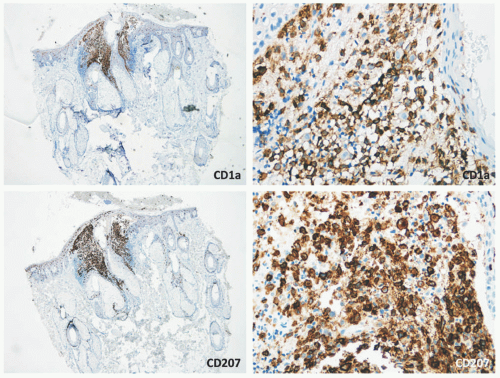 FIGURE 26-5. Langerhans cell histiocytosis—immunohistochemistry. The atypical Langerhans cells are positive for CD1a and CD207 (langerin). |
A careful distinction between reactive dermatoses with associated Langerhans cell hyperplasias and LCH is important to be made, in order to avoid overdiagnosing LCH. LCH typically fills the papillary and/or deeper dermis and sometimes can show a perifollicular and epidermal distribution. Chronic dermatoses often show increased number of perivascular and perifollicular dendritic cells that are CD1a+/langerin low to negative. Contact dermatitis shows frequent small intraepidermal clusters of Langerhans cells that lack significant cytologic atypia.36 Immunohistochemistry for cyclin D1 and p53 has also shown an adjunct role in distinguishing reactive (negative staining) Langerhans cell hyperplasias, from neoplastic (positive staining) forms.45,46
Differential Diagnosis
LCH is often discernable based on its characteristic cytology and unique immunohistochemical pattern. Non-neoplastic, reactive Langerhans cell hyperplasia secondary to an exogenous inflammatory insult such as an eczematous dermatitis, bug bites, or scabies should be considered. Non-LCH disorders such as JXG and ECD may
mimic LCH at various time points. Other entities that may be considered include lymphoma, LCS, malignant melanoma, and cutaneous T-cell lymphoma, most of which are easily differentiated based on immunohistochemistry. Melanoma can be ruled out based on staining for melanocytic markers. Also, melanoma usually starts in the epidermis and invades deeper tissues, whereas in LCH, the infiltrate centers in the papillary dermis, is associated with a mixed cell infiltrate, and spreads up to the epidermis via epidermotropism. ECD is a multisystem histiocytosis that often involves bone, but lesions contain foamy histiocytes and a different immunoprofile than that of LCH. JXG is characterized by foamy histiocytes and Touton giant cells, although the morphology of JXG varies based on the stage of the lesion (see Section “Juvenile Xanthogranuloma”).
mimic LCH at various time points. Other entities that may be considered include lymphoma, LCS, malignant melanoma, and cutaneous T-cell lymphoma, most of which are easily differentiated based on immunohistochemistry. Melanoma can be ruled out based on staining for melanocytic markers. Also, melanoma usually starts in the epidermis and invades deeper tissues, whereas in LCH, the infiltrate centers in the papillary dermis, is associated with a mixed cell infiltrate, and spreads up to the epidermis via epidermotropism. ECD is a multisystem histiocytosis that often involves bone, but lesions contain foamy histiocytes and a different immunoprofile than that of LCH. JXG is characterized by foamy histiocytes and Touton giant cells, although the morphology of JXG varies based on the stage of the lesion (see Section “Juvenile Xanthogranuloma”).
CAPSULE SUMMARY
LANGERHANS CELL HISTIOCYTOSIS
Today LCH is considered the prototypic histiocytosis from the “L” group. LCH cells are arranged in clusters and sheets in the papillary dermis and have abundant eosinophilic cytoplasm and irregular, reniform, or folded vesicular nuclei with longitudinal grooves. Epidermotropism may be a feature. By immunohistochemistry LCH shows the following phenotypes: CD1a+, S-100+, CD207/langerin+; CD68 is variable. Birbeck granules are present on electron microscopy, but CD207 is a surrogate marker.
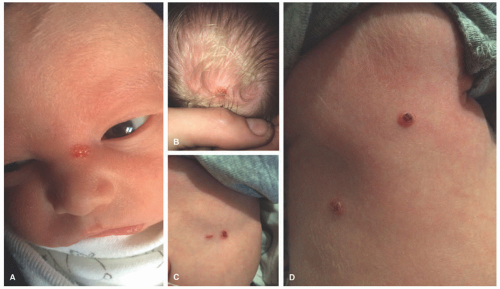 FIGURE 26-6. Congenital self-healing histiocytosis. Erosive red-tan papules in the nose (A), scalp (B), and trunk (C and D). |
CONGENITAL SELF-HEALING RETICULOHISTIOCYTOSIS (HASHIMOTO-PRITZKER DISEASE)
Definition and Epidemiology
CSHR (Hashimoto-Pritzker disease), initially described by Hashimoto and Pritzker in 1973, is generally considered to be a unique, self-limited variant of LCH that presents in the neonatal period and resolves spontaneously.47 CSHR is a disease mostly of neonates and sometimes in infants with no gender predilection.48 It is rarely reported, but may be underestimated because of its self-limited and often short-lived clinical course.49 The etiology is poorly understood, but it’s likely related to other common forms of LCH.
Clinical Presentation
CSHR presents at birth or in the neonatal period with an eruption of red-brown papules, nodules, or vesicles in a generalized distribution and can involve the mucosa (Figure 26-6).48 Rarely, CSHR presents as a solitary lesion. Secondary changes such as ulceration or crusting may be present. Cutaneous disease is typically self-limited, lasting weeks to months, and it rarely progresses to involve internal organs. It is difficult to predict when systemic involvement, relapse after cutaneous resolution, or a more aggressive clinical course will occur. There are no criteria for easily distinguishing CSHR from systemic LCH except for its clinical course; therefore, evaluation for systemic disease at the time of diagnosis and long-term follow-up are paramount.49
Poor prognostic indicators are later age of onset and/or the presence of systemic involvement.47,50,51
Poor prognostic indicators are later age of onset and/or the presence of systemic involvement.47,50,51
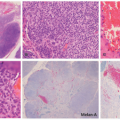
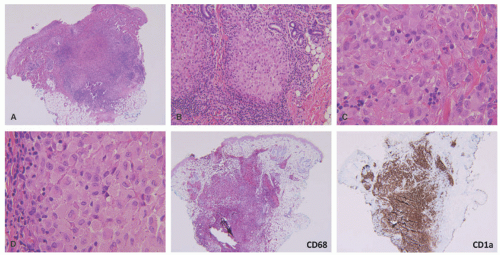 FIGURE 26-7. Congenital self-healing reticulohistiocytosis—histopathology. A, Deep dermal-based nodule with central areas of necrosis. B, The infiltrate has a nodular appearance. C and D, The infiltrate is composed of atypical Langerhans cells. The cells are positive for CD68 and CD1a. |
Histologic Findings
The histologic picture of CSHR is similar to that of LCH, and differentiation of the two entities is primarily clinical as they likely represent polar ends of a clinical spectrum of the same disease (Figure 26-7).47,52,53,54 In CSHR-LCH, lesions typically involve the deep dermis in a nodular fashion, unlike conventional LCH lesions where lesions are mainly in the upper dermis, extending to epidermis. Both clinical entities show similar staining with CD1a, S-100, and langerin. Langerhans cells of congenital self-healing LCH may show Birbeck granules similar to conventional LCH cells by electron microscopy; however, they can be separated ultrastructurally by the presence of unique cytoplasmic dense bodies containing concentrically arranged laminar structures. The differential diagnosis is similar to cases of ordinary LCH.
CAPSULE SUMMARY
CONGENITAL SELF-HEALING RETICULOHISTIOCYTOSIS (HASHIMOTO-PRITZKER DISEASE)
CSHR presents at birth or in the neonatal period with an eruption of red-brown papules, nodules, or vesicles in a generalized distribution and can involve the mucosa. Cutaneous disease is typically self-limited, lasting weeks to months, and it rarely progresses to involve internal organs. The histologic picture of CSHR is similar to that of LCH, and differentiation of the two entities is primarily clinical as they likely represent polar ends of a clinical spectrum of the same disease.
ERDHEIM-CHESTER DISEASE
Definition and Epidemiology
ECD, first reported by Erdheim and Chester as “lipid granulomatosis” in 1930, is a rare, multisystemic non-LCH that almost always affects bones, but can additionally involve almost any organ system.55 It is characterized by a disseminated xanthogranulomatous infiltration of organs with foamy histiocytes, progressive fibrosis, and ultimately organ failure.56 It’s now included under the group “L” of histiocytosis, given the characteristic molecular profile.
Etiology
The etiology has not been completely elucidated, but it is thought to be neoplastic in nature, representing a hematopoietic neoplasm of histiocytic origin, versus less
likely a purely reactive/inflammatory process. Recently, ECD has been associated with a mutation in the BRAF proto-oncogene in greater than 50% of cases, establishing that the disorder is clonal in nature and supporting a neoplastic process, perhaps with a shared origin to that of LCH.55,56,59,60 Additionally, mutations in the MAPK (such as in NRAS) and PIK3 pathways have been reported. Another finding in support of a Langerhans cell relationship is the fact that 20% of biopsies of ECD have coexistent LCH.1,61 However, these data were only based on small case series and case reports.
likely a purely reactive/inflammatory process. Recently, ECD has been associated with a mutation in the BRAF proto-oncogene in greater than 50% of cases, establishing that the disorder is clonal in nature and supporting a neoplastic process, perhaps with a shared origin to that of LCH.55,56,59,60 Additionally, mutations in the MAPK (such as in NRAS) and PIK3 pathways have been reported. Another finding in support of a Langerhans cell relationship is the fact that 20% of biopsies of ECD have coexistent LCH.1,61 However, these data were only based on small case series and case reports.
Clinical Presentation
The disease is diagnosed most often between 40 and 60 years of age, and bone pain accompanied by fatigue is the most common presentation, with the skeletal system being involved in 95% of cases of ECD. Pathognomonic radiographic features are bilateral, symmetric, and multifocal osteosclerotic changes of the diametaphyseal regions of long bones sparing the epiphyses. This is in contrast to the lytic lesions seen in LCH, which typically affect the skull and not the long bones.60,62 Diagnosis is based on a combination of clinical, histologic, and radiographic features. Skin involvement, which occurs in up to a third of cases, typically includes xanthoma-like papules and periorbital xanthelasma-like plaques, and less commonly red-brown papular lesions of the trunk and extremities (Figure 26-8).57,60,63 In ECD, the periorbital plaques are often more nodular than xanthelasma and may extend into the temporal region, cheeks, or subpalpebral areas—such irregularities in addition to a normal patient lipid profile should raise the consideration that the lesions may be more than simple xanthelasma.
Clinical presentations of ECD vary broadly based on which organ systems are involved and can include: exophthalmos, papilledema, papulonodular skin lesions, endocrine dysfunction (from pituitary involvement), diabetes insipidus, lung disease, retroperitoneal fibrosis, renal failure, cardiomyopathy, and CNS disorder.55,59 Almost all patients have extraskeletal involvement, with cardiac and neurologic involvement harboring the worst prognosis. 62 Bilateral, symmetric sclerosing bone lesions in the metaphysis of long bones, usually the distal femur and proximal tibia and fibula, are the diagnostic imaging features.13,24,25,57,64 The 3-year mortality rate from time of diagnosis is estimated to be about 60%, and the most common causes of death include respiratory or cardiac failure.56,65
Histologic Findings
The histology of ECD, which is not entirely specific, shows a dense dermal infiltrate of bland, foamy histiocytes with variable amounts of intervening fibrosis, Touton giant cells, and mixed inflammatory cells such as plasma cells and lymphocytes.66 Biopsy from involved internal sites yields comparable histologic findings (Figures 26-8 and 26-9). Cytological atypia is not a feature. ECD shares resemblance with other non-LCH such as progressed JXG, where fibrosis becomes a predominant feature. The immunohistochemical profile of the foamy macrophages is positive for CD68, CD163, CD14, and factor XIIIa and negative for S-100, CD1a, and CD207.59 ECD-associated xanthelasma-like plaques have more multinucleate and Touton giant cells and have less fibrosis than common xanthelasma. When encountering xanthelasma-like lesions, testing for BRAF mutation is indicated when there is a high clinical suspicion for ECD.63,67,68 In children, ECD can occur in association with lymphoblastic leukemias and LCH.57,58,69,70 ECD can also have a presentation that mimics multicentric reticulohistiocytosis (MRH).71
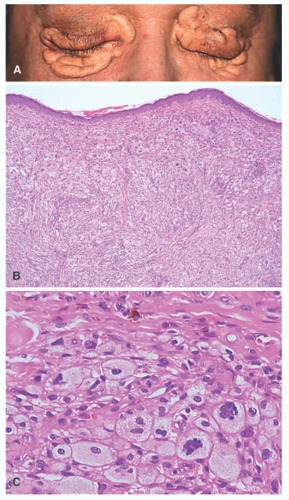 FIGURE 26-8. Erdheim-Chester disease (ECD). Xanthelasma-like tumors as cutaneous ECD manifestations. A, Large xanthelasma-like tumor around the eyes. B, Diffuse accumulation of foamy cells, fibrosis of the connective tissue (hematoxylin and eosin ×40). C, Multiple Touton giant cell and hemosiderin deposits in the dermis (hematoxylin and eosin ×400). Obtained with permission. Liersch J, Carlson JA, Schaller J. Histopathological and clinical findings in cutaneous manifestation of Erdheim-Chester disease and Langerhans cell histiocytosis overlap syndrome associated with the BRAFV600E mutation. Am J Dermatopathol. 2017;39(7):493-503. doi:10.1097/DAD.0000000000000793. |
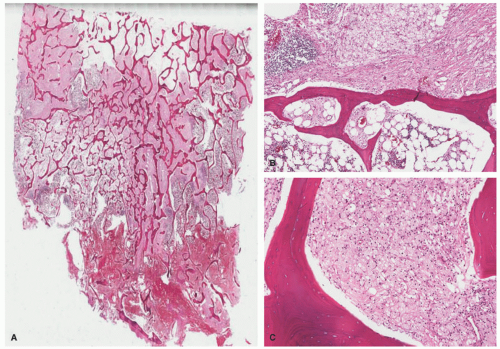 FIGURE 26-9. Erdheim-Chester disease (ECD). This bone marrow biopsy shows the classic morphologic findings of ECD involvement. A and B, There is a diffuse histiocytic infiltrate replacing most of the bone marrow space. C, The infiltrate is composed of foamy histiocytes and scattered multinucleated giant cells. |
Differential Diagnosis
The clinical differential diagnosis includes other systemic diseases, such as systemic JXG, xanthoma disseminatum (XD), RDD, LCH, sarcoidosis, amyloidosis, and storage disorders. The histiocytic cells of ECD are positive for CD163 and CD68, whereas being negative for CD1a and langerin. Some cases can have partial expression of S-100. Additionally, a recent study has shown expression of the programmed cell death ligand 1 (PD-L1) protein in a subset of histiocytoses, including ECD disease, suggesting a possible role of immune checkpoint blockade in treatment. Rare cases with histologic and immunophenotypic features of ECD, but with strong ALK expression and rearrangement of the ALK gene, can be seen. Such cases are examples of ALK+ histiocytosis of childhood and not ECD.72 ECD is often progressive and fatal despite aggressive therapy. The recent identification of various mutations in ECD has resulted in some success with targeted therapies, such as with vemurafenib. In a recent pangenomic analysis, all cases of ECD had at least one mutation activating the MAPK pathway.23 In addition, in-frame fusions involving several kinases including BRAF, ALK, and NTRK1 were identified in ECD without BRAF, MAP2K1, or N/KRAS point mutations.13,24,25 ECD should also be distinguished from JXG. As opposed to JXG, ECD more frequently harbors the BRAF V600E mutation. ECD also has the typical clinical and radiographic findings, not present in cases of JXG. The recently reported cases of BRAFmutated JXG are now believed to be examples of ECD.73,74
CAPSULE SUMMARY
ERDHEIM-CHESTER DISEASE
ECD is a rare, multisystemic non-LCH that almost always affects bones, but can additionally involve almost any organ system. It is characterized by a disseminated xanthogranulomatous infiltration of organs with foamy histiocytes, progressive fibrosis, and ultimately organ failure. It is now included under the group “L” of histiocytosis, given the characteristic molecular profile. The histopathology of ECD is reminiscent of other non-LCH. There is a dense dermal infiltrate of foamy histiocytes with variable amounts of intervening fibrosis. Touton giant cells and mixed inflammatory cells are also seen. It is important to consider testing for BRAF mutation to help distinguish from xanthelasma if clinical suspicion exists.
INDETERMINATE DENDRITIC CELL TUMOR
Definition and Epidemiology
Indeterminate dendritic cell tumor (IDCT) is a rare histiocytic proliferative disorder that is thought to originate from dermal indeterminate histiocytes that typically share the immunophenotype of macrophages (CD68+) and Langerhans cells (expressing S-100 and CD1a). Indeterminate cells lack Birbeck granules—the ultrastructural pathognomonic feature of Langerhans cells—and they are nonreactive with antibodies to langerin (also known as CD207, an immunohistochemical marker that indicates the presence of Birbeck granules).75 It was first reported by Wood et al in 1985.76 IDCT is very rare (less than 50 reported cases), making the epidemiology and demographics difficult to assess. It can occur at any age, including childhood, but appears to be more prevalent in adults, and it has no gender predilection.75,77,78,79,80 The updated revision of cutaneous histiocytoses includes IDCT in the “L” (“Langerhans”) group.
Etiology
The cause of IDCT is unknown. Some consider the indeterminate cell to be an immature Langerhans cell that has not yet developed Birbeck granules.76 It has been reported in association with hematologic malignancies such as B-cell lymphoma.81 It has also been reported to be triggered by pityriasis rosea and scabies.79 Recently, Brown et al described three cases of IDCT with an ETV3-NCOA2 translocation.82 That abnormality has not been identified in other histiocytic lesions, and it provides solid evidence that some IDCTs do indeed represent a neoplasm with a recurrent molecular aberration.
Clinical Presentation
IDCT is mostly a disease of the skin and usually presents as either solitary or multiple pink to red papules and nodules on the trunk, extremities, and/or head and neck, sparing the mucosa. The lesions are typically asymptomatic and may either be generalized or focal in distribution. Extracutaneous manifestations that have been reported include nodal, corneal, osseous, and genital involvement (Figure 26-10). ICH usually engenders a benign clinical course, especially when not associated with systemic illness such as hematologic malignancy, and it resolves spontaneously without treatment.75,81
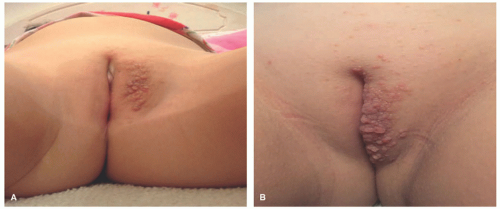 FIGURE 26-10. Indeterminate dendritic cell tumor. A and B, Multiple papules in the vulvar area. |
Histologic Findings
The histologic picture of IDCT is that of a dense dermal infiltrate composed of large, monomorphous mononuclear cells with vesicular nuclei (occasionally reniform) and abundant pale cytoplasm.83 Cases with a spindle cell morphology have been reported (Figure 26-11).78,84 Multinucleate (Touton) and/or xanthomatized cells may be seen.85,86 There may be scattered inflammatory cells present as well.76 Epidermotropism is typical of ICH, but mitoses may be seen. Immunophenotype: CD1a+, S-100+, CD68+, langerin (CD207), Birbeck granules (Figure 26-12). An S-100-negative variant has been reported.77
Differential Diagnosis
The most important entity on the differential diagnosis is LCH, which, unlike IDCT, displays positivity for CD207 (langerin) and contains Birbeck granules. CD207 (langerin) is an immunohistochemical stain for a protein found in Birbeck granules and can be used in lieu of electron microscopy. Furthermore, in LCH, there is often epidermotropism, intercellular edema, and clinically internal involvement. Up to 50% of cases of LCH have BRAF V600E mutations, a feature that is much less prevalent in IDCT. Non-LCH such as JXG and generalized eruptive histiocytoma often resemble IDCT, both clinically and histologically, but the
immunohistochemical profile of the disorders is distinct, with ICH demonstrating positivity for S-100 and CD1a, both of which are negative in the non-LCH disorders.
immunohistochemical profile of the disorders is distinct, with ICH demonstrating positivity for S-100 and CD1a, both of which are negative in the non-LCH disorders.
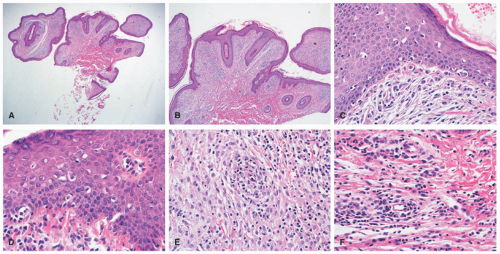 FIGURE 26-11. Indeterminate dendritic cell tumor—histopathologic findings. A and B, In this case, there is a dermal-based histiocytic proliferation accompanied by epidermal acanthosis and papillomatosis. C and D, The infiltrate shows epidermotropism. Within the dermis, the atypical histiocytes show a spindle cell appearance (E), and scattered eosinophils are seen (F). |
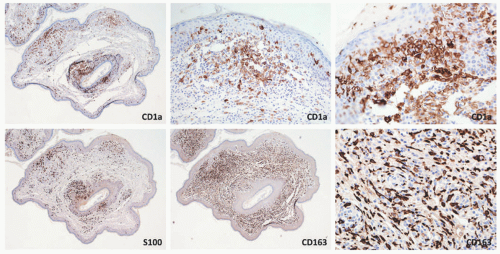 FIGURE 26-12. Indeterminate dendritic cell tumor—immunohistochemistry. The epidermotropic and dermal histiocytic cells are positive for CD1a, S-100, and CD163. |
CAPSULE SUMMARY
INDETERMINATE DENDRITIC CELL TUMOR
IDCT is a rare histiocytic proliferative disorder that is thought to originate from dermal indeterminate histiocytes that typically share the immunophenotype of macrophages (CD68+) and Langerhans cells (expressing S-100 and CD1a). The lesional cells are langerin negative (CD207) and show absence of Birbeck granules. The ETV3-NCOA2 translocation has been demonstrated in a subset of cases.
GROUP “C” HISTIOCYTOSES
JUVENILE XANTHOGRANULOMA
Definition and Epidemiology
JXG (xanthogranuloma, nevoxanthoendothelioma) is a self-limited non-LCH involving proliferation of cholesterol-laden monocyte-derived macrophages that most commonly occur in the first year of life.87 Although JXG are typically benign and confined to the skin, rarely, they may involve extracutaneous sites or be associated with other disease processes.88 JXG is the most common form of non-LCH and is currently classified as group “C” for the cutaneous limited forms and in the “L” category for the systemic variants.1 The exact incidence is unknown as most cases are diagnosed based upon clinical findings and without histologic confirmation. One study reported that JXG represents only 0.5% of pediatric tumors. Most cases are diagnosed in infants and young children, often in the first year or two of life.89,90,91 Males are slightly affected more often than females, especially when lesions are multiple. Less commonly, JXG occurs in adolescents and young adults and in some cases may be difficult to differentiate from ECD, except for the BRAF V600E mutations.
Although primarily a disease of childhood, it may occur in adults as well. It is slightly more common in boys (1.5:1) and is more often reported in Caucasians. Given their transient and classically benign nature, they are likely underdiagnosed, especially by nondermatology physicians. Furthermore, JXG often presents as a small, solitary lesion that may easily be mistaken for a common benign melanocytic nevus.92
Etiology
The pathogenesis of JXG is poorly understood.91 It has been postulated to represent a reactive process to an unknown stimulus or injury, but this has not been verified in studies.92,93 There may be genetic mutations that play a role in the pathogenesis of JXG, such as that seen in several other histiocytoses like LCH and ECD, and this is currently being studied.75,94
JXG can be associated with neurofibromatosis type 1 (NF1) and juvenile myelomonocytic leukemia (JMML). Nearly 5% to 10% of NF1 patients and up to 30% of NF1 patients less than 2 years of age have been reported to have JXG. The presence of café au lait macules and JXG should suggest the likelihood of NF1. The association among JXG, NF1, and JMML, while well described, is poorly understood in terms of the pathogenetic relationships. JXG usually precedes JMML; however, it is unclear if JXG is a reliable marker for the subsequent development of JMML in NF1 patients. It has been suggested that mutations in the GTPase neurofibromin, which is present in NF1, lead to RAS dysfunction and downregulation of Fas antigen, with subsequent dysregulation of apoptosis in hematopoietic cells with the development of leukemia and JXG. Some cases of JXG follow the diagnosis of LCH.95,96,97,98,99,100,101,102 More recently, systemic presentations of JXG with CNS involvement have shown BRAF V600E mutations.73,74 The question that arises from these is: should those cases be qualified as ECD/L group lesions that haven’t presented themselves with systemic findings (ie, brain only) or is there a truly separate group of JXG/BRAF V600E-positive tumors?
Clinical Presentation
JXG often presents very early in life. Indeed, about 10% are present at birth and up to 70% occur within the first year of life. They rarely occur in the adult population, and they are not associated with lipid abnormalities. The classic presentation of JXG is the abrupt onset of a firm, painless, and freely mobile papulonodule (Figure 26-13). JXG appears as a solitary nodule in 60% to 80% of cases, and the head and neck are the most common locations. Less common cutaneous sites include the genitalia, soles, and fingers.64,102,103,104 The shape of the lesion can vary greatly, as it may be keratotic, pedunculated, linear, flat, or plaque like. The papules or nodules are usually 5 to 10 mm in diameter but can measure up to a few centimeters.105,106,107 Initially, the lesion is raised and pink to erythematous in color, but over time, they become yellow-brown and may flatten as the histiocytes undergo xanthomatous transformation and regression (Figure 26-14). Regression may occur over a period of months to years with residual hyperpigmentation, atrophy, or anetoderma. Telangiectatic blood vessels or ulceration may be seen overlying the lesion, which imparts a resemblance to pyogenic granuloma. Multiple skin lesions occur infrequently, in less than 10% of cases.89 JXG is mostly asymptomatic, but on occasion may be pruritic, painful, and/or ulcerate. The characteristic finding on dermoscopy is the “setting sun” pattern in which the center of the lesion is yellow surrounded by a circumferential orange-red rim.108
Although JXG is typically limited to the skin in the majority of cases, it may arise in extracutaneous sites with or without skin involvement.
Although JXG is typically limited to the skin in the majority of cases, it may arise in extracutaneous sites with or without skin involvement.
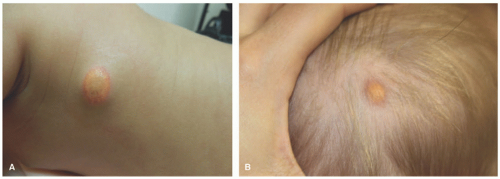 FIGURE 26-13. Juvenile xanthogranuloma. Mature lesions: xanthomatized plaques on the trunk (A) and scalp (B). |
JXG often resolves over the course of several years and may leave behind dyspigmentation or, more rarely, anetoderma, atrophy, or scarring.92 Extracutaneous disease occurs in about 4% of patients, and the eye (iris followed by the eyelid) is the most common location with an overall incidence of 0.3% to 0.5%. The next most common extracutaneous sites of involvement are the lung and the liver.75,109 Ocular involvement is usually accompanied by multiple cutaneous lesions, and complications of ocular JXG include hyphema, glaucoma, uveitis, and blindness.91,92 CNS involvement is very rare, but is associated with poor outcomes. The prognosis for exclusively cutaneous JXG is excellent; however, if there is systemic involvement, mortality ranges from 2% to 20%.75
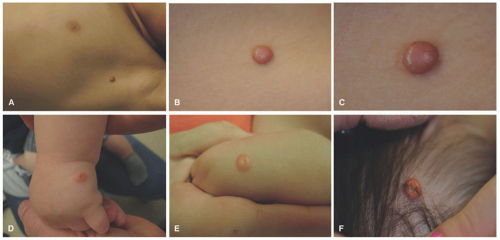 FIGURE 26-14. Juvenile xanthogranuloma. A to C, Early lesions show red-brown papules. D to F, Mature lesions have a yellow color, given the number of xanthomatized histiocytes. |
Histologic Findings
A dense mononuclear histiocytic infiltrate in the dermis extending to the interface with the epidermis is present.89,110,111 In smaller lesions, the infiltrate may be limited to the superficial dermis but, in larger lesions, may extend into the subcutis. There is often flattening of the epidermis with loss of the rete ridges; however, the epidermis and adnexa are spared. The infiltrate is composed of small mononuclear cells, as well as multinucleated giant cells with and
without the features of Touton giant cells (Figures 26-15 and 26-16). In fact, cutaneous JXG may be devoid of giant cells, and their presence in extracutaneous lesion is often difficult to demonstrate. Lymphocytes, eosinophils, plasma cells, neutrophils, and mast cells may be scattered throughout the infiltrate or be altogether absent. The morphology of the histiocytes can vary depending on the “age” of the lesion that is often difficult to determine with any certainty. So-called early lesions are composed predominantly of mononuclear cells with abundant eosinophilic and minimally vacuolated cytoplasm. More “mature” lesions contain histiocytes with “lipidized or xanthomatized” cytoplasm with numerous fine cytoplasmic vacuoles. Touton giant cells, which are characterized by a ring or wreath of nuclei surrounded by a rim of foamy cytoplasm, are characteristic, but their presence is variable in this phase. Scalloped histiocytes with an angulated or jagged border and spindled histiocytes may also be seen. In fact, some lesions are composed predominantly of spindle cells (Figure 26-17), but the finely vacuolated interspersed mononuclear cells are the clue to the diagnosis of JXG. Not entirely surprising is the resemblance in some cases between JXG and dermatofibroma (DF). Mitotic figures may be seen, but atypical mitotic figures are absent.
without the features of Touton giant cells (Figures 26-15 and 26-16). In fact, cutaneous JXG may be devoid of giant cells, and their presence in extracutaneous lesion is often difficult to demonstrate. Lymphocytes, eosinophils, plasma cells, neutrophils, and mast cells may be scattered throughout the infiltrate or be altogether absent. The morphology of the histiocytes can vary depending on the “age” of the lesion that is often difficult to determine with any certainty. So-called early lesions are composed predominantly of mononuclear cells with abundant eosinophilic and minimally vacuolated cytoplasm. More “mature” lesions contain histiocytes with “lipidized or xanthomatized” cytoplasm with numerous fine cytoplasmic vacuoles. Touton giant cells, which are characterized by a ring or wreath of nuclei surrounded by a rim of foamy cytoplasm, are characteristic, but their presence is variable in this phase. Scalloped histiocytes with an angulated or jagged border and spindled histiocytes may also be seen. In fact, some lesions are composed predominantly of spindle cells (Figure 26-17), but the finely vacuolated interspersed mononuclear cells are the clue to the diagnosis of JXG. Not entirely surprising is the resemblance in some cases between JXG and dermatofibroma (DF). Mitotic figures may be seen, but atypical mitotic figures are absent.
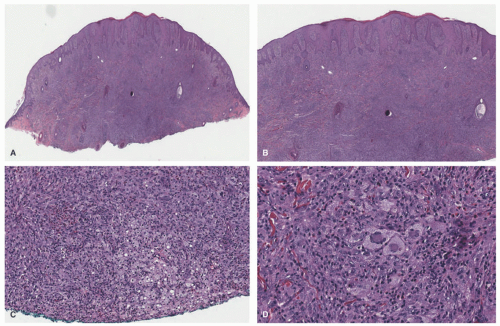 FIGURE 26-15. Juvenile xanthogranuloma—histopathologic findings. A and B, Dense dermal histiocytic infiltrate with epidermal hyperplasia. C, In this early lesion, only focal xanthomatized changes are seen. D, Scattered Touton-type giant cells are seen. Digital slides courtesy of Path Presenter.com. |
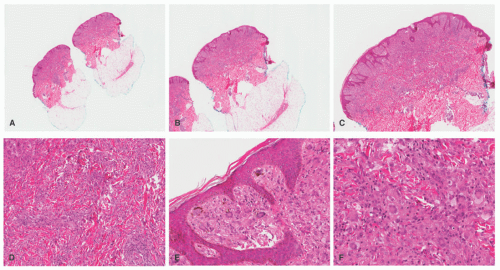 FIGURE 26-16. Juvenile xanthogranuloma—histopathologic findings. A to F, This is a relatively more mature lesion, with a larger proportion of xanthomatized histiocytes and giant cells. |
The histiocytes stain positively for CD68, CD163, CD4, CD14, factor XIIIa, HLA-DR, fascin, vimentin, and lysozyme and are negative for CD1a and langerin.89,111 The histiocytes are usually negative for S-100 but it is not unusual to encounter individual and patchy staining in some cases. Frequent admixed S-100 dendritic cells can be seen in some cases. Ultrastructural examination of “mature” lesions shows lipid vacuoles, lysosomes, cholesterol clefts, comma-shaped bodies, and myeloid bodies in the cytoplasm of the histiocytes. No Birbeck granules are present.
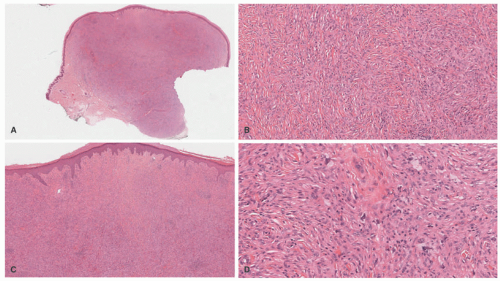 FIGURE 26-17. Juvenile xanthogranuloma—spindle cell variant. This form is a potential mimicker of dermatofibromas (DF). The lesion lacks the surface induction changes seen in DF (A and B) or the collagen “trapping” at the periphery of the lesions. The infiltrate is composed of spindle cells with admixed xanthomatized cells and Touton-type giant cells (C and D). Digital slides courtesy of Path Presenter.com. |
Differential Diagnosis
The histologic differential diagnosis of JXG varies based on the stage of the lesion, but may include reticulohistiocytoma, benign fibrous histiocytoma (DF), LCH, Spitz nevus, melanoma, and malignant fibrous histiocytoma. In early JXG in which Touton giant cells and xanthomatized histiocytes are absent, differentiating JXG from LCH may be difficult. In such cases, staining for S-100 and CD1a, both of which are positive in LCH and negative in JXG, may be used to make the diagnosis.
The lipidized variant of DF contains numerous large, foamy cells and Touton-like giant cells, reminiscent of JXG. However, areas of spindle cells in a whorled or storiform pattern may be present, and collagen trapping can be seen at the periphery of the lesion. However, the distinction between JXG and DF in an older child is a challenge, with some degree of uncertainty. DFs tend to have overlying epidermal changes, such as acanthosis, hyperpigmentation of epidermal basal cells, or folliculosebaceous induction, and often contain hemosiderin unlike JXG. Both lesions can
be positive for factor XIIIa, but DFs are usually negative or only weakly positive for CD68.
be positive for factor XIIIa, but DFs are usually negative or only weakly positive for CD68.
Reticulohistiocytoma can also mimic JXG clinically and histologically. The histopathologic findings of reticulohistiocytoma, including the presence of densely eosinophilic “ground glass” cytoplasm, usually allow for a distinction from JXG; however, there may be immunohistochemical overlap between these lesions as the reticulohistiocytomas are also positive for CD68 and CD163, negative for CD1a, and often negative for S-100. It should be emphasized that some consider reticulohistiocytomas as histologic variants of JXG.1,36,112 Spitz nevi can also enter the differential diagnosis; however, the melanocytic nature of the lesion can readily be demonstrated with the application of the melanocytic markers, Melan-A, HMB-45, or SOX-10. Mast cell neoplasms (mastocytomas) can also resemble JXG (when the latter are devoid of lipidized cells). Mastocytomas are characteristically positive for CD117 and tryptase and have aberrant coexpression of CD2 and CD25. α-Chloroacetate esterase (Leder stain) is positive in the mast cell granules. Systemic JXG should also be distinguished from ALK+ histiocytosis of childhood. ALK+ histiocytosis manifests with anemia, massive hepatosplenomegaly, and thrombocytopenia. The liver shows sinusoidal infiltration by histiocytic cells with folded nuclei, fine chromatin, small nucleoli, and voluminous eosinophilic cytoplasm with occasional hemophagocytosis.72 Therefore, when diagnosing ECD, ALK immunostain should always be performed.
CAPSULE SUMMARY
JUVENILE XANTHOGRANULOMA
JXG (xanthogranuloma, nevoxanthoendothelioma) is a self-limited non-LCH involving proliferation of cholesterol-laden monocyte-derived macrophages that most commonly occurs in the first year of life. It is the most common form of non-LCH and is currently classified as group “C” for the cutaneous limited forms and in the “L” category for the systemic variants. The infiltrate is composed of small mononuclear cells, as well as multinucleated giant cells with and without the features of Touton giant cells.
BENIGN CEPHALIC HISTIOCYTOSIS
Definition and Epidemiology
First described by Gianotti et al in 1971, BCH is an uncommon non-LCH that occurs in young children and presents as a self-healing, asymptomatic eruption of papules on the head and neck. It is benign in nature and is not associated with systemic disease or visceral involvement. BCH likely lies on a spectrum of non-LCH disorders such as Generalized eruptive histiocytosis (GEH) and JXG.113,114 They are included under group “C.”
BCH is a rare disorder that presents in infants and young children. The average age of onset is 15 months, and up to 45% of cases occur in patients less than 6 months of age. No gender predilection has been observed.114 About 60 cases have been reported in the English literature, but the disorder may be underreported.115,116
Etiology
Clinical Presentation
BCH is typified by an eruption of brown to tan, flattopped papules that range in size from about 1 to 8 mm (Figure 26-18). This condition almost always starts on the face, after which it may spread to involve the trunk and/or extremities. It does not involve mucosal or acral sites and has not been reported to affect internal organs, except in one case that was associated with infiltration of the pituitary stalk leading to diabetes insipidus.119,120 The total number of lesions can range from as few as 5 to as many as 100. Lesions typically regress over the course of several years and can leave behind hyperpigmentation, but usually do not scar.114,121
Histologic Findings
The histology of BCH is characterized by a well-defined proliferation of histiocytes with oval to reniform nuclei and abundant eosinophilic cytoplasm (Figure 26-19). The infiltrate lies in the mid-upper dermis adjacent to, but sparing the epidermis, which may appear flat and atrophic. The infiltrate is often accompanied by a mixed inflammatory cell infiltrate. Xanthomatous change can occur as lesions evolve and scattered multinucleate giant cells may be present. Such findings are identical to those seen in JXG. Immunostaining of the infiltrate reveals positivity for CD68, CD163, HAM-56, and factor XIIIa and negativity for CD1a, CD 207 (langerin), and usually S-100.117,122 Comma-shaped bodies may be seen on ultrastructural evaluation via electron microscopy, but in contrast to LCH, Birbeck granules are invariably absent.114,118
Differential Diagnosis
In general, the histologic differential diagnosis is the same as for JXG, and it most importantly includes LCH, which is S-100 and CD1a positive. Other non-LCH disorders such as JXG and GEH are primarily distinguished based on clinical features, as the histopathology alone is undifferentiating, especially in the early nonxanthomatous phase.116
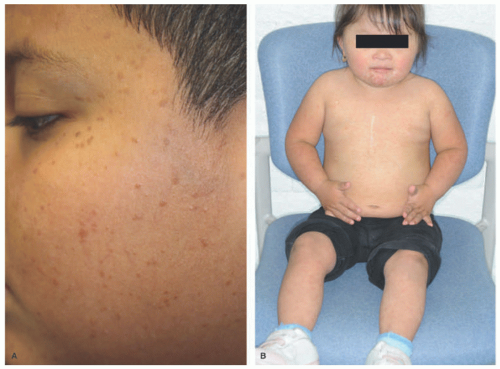 FIGURE 26-18. Benign cephalic histiocytosis—two examples. Small, brown-tan papules in the face (A and B). |
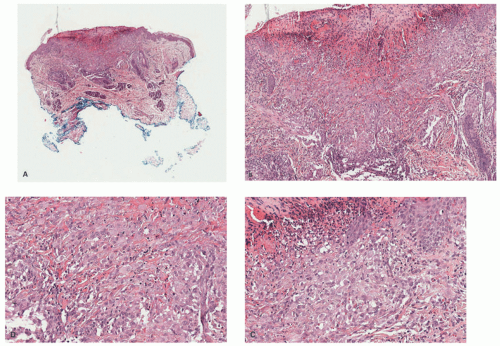 FIGURE 26-19. Benign cephalic histiocytosis—histopathologic findings. The biopsy shows an ulcerated epidermis (A) with a dermal-based infiltrate (B). The lesion is composed of epithelioid histiocytes and scattered multinucleated giant cells (C and D). Digital slides courtesy of Path Presenter.com. |
CAPSULE SUMMARY
BENIGN CEPHALIC HISTIOCYTOSIS
Similar to JXG, BCH demonstrates a dermal CD68+ histiocytic infiltrate. BCH is histologically indistinguishable from other non-LCH such as JXG and GEH. The evolution of the infiltrate is marked by xanthomatization followed by fibrosis. Comma-shaped bodies can be seen on electron microscopy.
PROGRESSIVE NODULAR HISTIOCYTOSIS
Definition and Epidemiology
PNH, first described in 1978, lies on the spectrum of non-LCH most closely resembling JXG. However, unlike JXG, the skin lesions have a chronic, progressive course only rarely undergoing spontaneous resolution.123,124 PNH is rarely reported, and there is a high degree of variance regarding the nomenclature of this disorder.
Etiology
Like the other closely related and overlapping “C” group of disorders, PNH is a proliferative disorder of dermal dendrocytes with an unknown cause. It is most similar to that of JXG, although it presents later and does not spontaneously regress over time.123,125,126 It is possible that PNH is not a distinct entity and merely represents a distinct clinical presentation of JXG.127
Clinical Presentation
Clinically, PNH is characterized by the progressive appearance of asymptomatic persistent, yellow to brown superficial papules and deep nodules on the skin and rarely mucosa that enlarge over time and can lead to significant physical distortion (Figure 26-20).125,128 The face is commonly involved and may progress to become leonine in appearance.129 There have been various reports of PNH occurring in patients with systemic illness such as blood cancers and hypothalamic tumors, but the precise relationship between PNH and the underlying illness was unknown.123,125,130 PNH has been reported in association with laryngeal lesions leading to respiratory failure and death.131
Histologic Findings
The histology of PNH closely resembles that of JXG with a dense dermal infiltrate of histiocytes with large, vacuolated cytoplasm and occasional multinucleated giant cells (Figure 26-21). PNH shows a tendency toward spindled histiocytes arranged in a storiform pattern (similar to spindle cell xanthogranuloma). Scattered inflammatory cells, xanthomatized histiocytes, and variable Touton giant cells may be seen.129,132 Similar to JXG, fibrosis becomes a prominent feature over time. The immunoprofile is identical to that of JXG, that is, positive for CD68 and HAM56, and negative for S-100 and CD1a.123,125
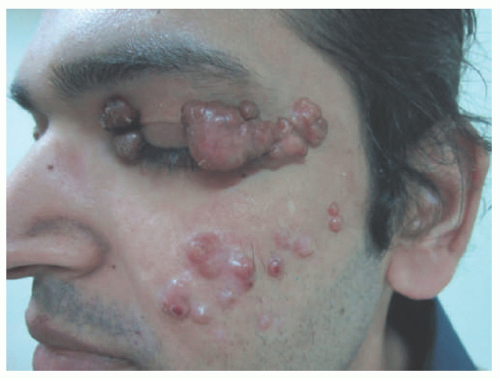 FIGURE 26-20. Progressive nodular histiocytosis. Papular and nodular lesions on the face and eyelid. Obtained with permission. Williams A, Thomas AG, Kwatra KS, Jain K. Progressive nodular histiocytosis associated with Eale’s disease. Indian J Dermatol. 2015;60(4):388-390. doi:10.4103/0019-5154.160492. |
Differential Diagnosis
See the differential diagnosis for JXG. Lesions with prominent storiform architecture may also demonstrate collagen trapping at the periphery of the infiltrate reminiscent of DF; however, PNH has a different immunoprofile and does not have overlying epidermal changes similar to that of DF.133
CAPSULE SUMMARY
PROGRESSIVE NODULAR HISTIOCYTOSIS
The histology of PNH closely resembles JXG and shares the same immunoprofile. JXG and PNH are primarily distinguished clinically.
XANTHOMA DISSEMINATUM
Definition and Epidemiology
XD (Montgomery syndrome) is a systemic, mucocutaneous form of proliferative histiocytic disease of histiocytic origin that primarily involves the skin, respiratory tract, and mucosal sites. Unlike most forms of non-LCH, XD can cause substantial morbidity and mortality because of airway involvement
and occasional internal disease, particularly that of the bone and the CNS.134,135,136 XD is very rare with around 100 reported cases. It can occur at any age, but most commonly occurs in young males, with a male to female ratio of 2.4:1.137 Reportedly, 60% of cases occur between the ages of 5 and 25 years.134
and occasional internal disease, particularly that of the bone and the CNS.134,135,136 XD is very rare with around 100 reported cases. It can occur at any age, but most commonly occurs in young males, with a male to female ratio of 2.4:1.137 Reportedly, 60% of cases occur between the ages of 5 and 25 years.134
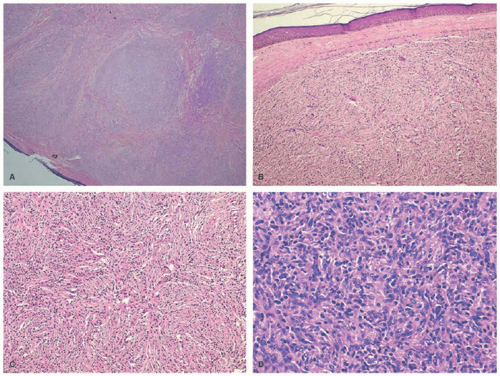 FIGURE 26-21. A, Dermal tumor with a nodular and diffuse growth pattern. Hematoxylin and eosin (H&E) ×20. B, Spindle-shaped tumor cells arranged in short fascicles. H&E ×40. C, Storiform pattern. H&E ×100. D, Histiocytes admixed with lymphocytes. H&E ×400. Obtained with permission. Williams A, Thomas AG, Kwatra KS, Jain K. Progressive nodular histiocytosis associated with Eale’s disease. Indian J Dermatol. 2015;60(4):388-390. doi:10.4103/0019-5154.160492. |
Etiology
XD is a sporadic disorder and unlike most xanthomatoses, patients with XD typically are normolipidemic, with up to 20% of patients having lipid abnormalities. The cause of XD is unknown, but it is currently thought of as a reactive proliferative disorder of macrophages to an unknown antigen. Over time, the histiocytes secondarily accumulate lipid either because of increased production and increased uptake or because of decreased efflux.135,137,138,139
Clinical Presentation
Clinically, XD is characterized by a progressive eruption of numerous (often hundreds) asymptomatic yellow-brown papules, plaques, and nodules that typically first appear symmetrically in the axilla or other intertriginous sites (Figure 26-22). The eruption then spreads to the face, especially the periocular region, as well as to the trunk and extremities. XD involves the mucous membranes 40% to 60% of the time with a predilection for the respiratory tract.134,137,140 The natural history of XD is most commonly that of persistent cutaneous disease or, less often, a progressive form with systemic involvement. The regressive type is the least common presentation and occurs in about a third of patients.136 XD has been associated with the following systemic sequela: osteolytic bone lesions, myeloma, Waldenstrom macroglobulinemia, monoclonal gammopathy, hyper- or hypothyroidism, diabetes insipidus from infiltration of the pituitary stalk, and neurologic disorders (epilepsy, cerebellar ataxia, hydrocephalus).137 XD with CNS involvement is rare, but when it occurs it may have a poor prognosis depending on which structures are involved.136 The clinical findings of XD can be
summarized as a triad of widespread normolipidemic xanthoma, mucous membrane involvement of the upper respiratory tract, and transient diabetes insipidus in approximately up to 40% of cases, though this triad does not occur in every patient.114,140,141,142
summarized as a triad of widespread normolipidemic xanthoma, mucous membrane involvement of the upper respiratory tract, and transient diabetes insipidus in approximately up to 40% of cases, though this triad does not occur in every patient.114,140,141,142
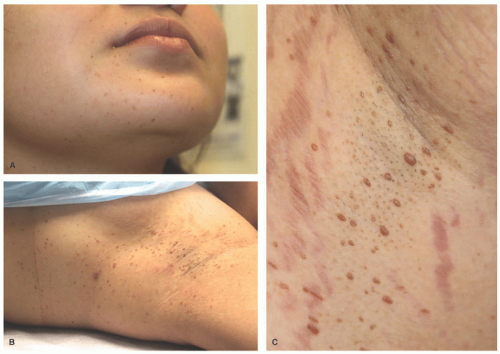 FIGURE 26-22. Xanthoma disseminatum. A to C, Numerous yellow-brown papules, plaques, and nodules that typically first appear symmetrically in the axilla or other intertriginous sites. |
Histologic Findings
The histology of XD overlaps with that of the other non-LCH disorders. A diffuse dermal infiltrate of large, pleomorphic scalloped histiocytes predominates in early lesions and over time Touton and foreign body giant cells, foamy macrophages, sparse inflammatory cells, and spindled cells begin to appear. The immunoprofile of XD is as follows: CD68+, factor XIIIa+, S-100–, and CD1a–.134,137,143
Differential Diagnosis
See the differential diagnosis for JXG. XD is differentiated from other non-LCH primarily on clinical grounds given the histologic overlap. Immunohistochemistry can rule out LCH.
CAPSULE SUMMARY
XANTHOMA DISSEMINATUM
XD (Montgomery syndrome) is a systemic, mucocutaneous form of proliferative histiocytic disease of histiocytic origin that primarily involves the skin, respiratory tract, and mucosal sites. Clinically, XD is characterized by a progressive eruption of numerous (often hundreds) asymptomatic yellow-brown papules, plaques, and nodules that typically first appear symmetrically in the axilla or other intertriginous sites. The histology of XD overlaps with that of the other non-LCH disorders.
SEA-BLUE HISTIOCYTE SYNDROME
Definition and Epidemiology
In 1970, sea-blue histiocyte syndrome (SBHS, OMIM 269600) was described by Silverstein as a disorder in which sea-blue histiocytes occupy various organs, especially the
bone marrow, liver, and spleen.144 The sea-blue histiocyte is a large macrophage that contains a cytoplasm filled with granules that turn blue-green in color when stained with Giemsa. It is now recognized that the disorder may either be inherited or represent a secondary phenomenon because of an underlying systemic illness. SBHS is exceptionally rare with about 60 reported cases.
bone marrow, liver, and spleen.144 The sea-blue histiocyte is a large macrophage that contains a cytoplasm filled with granules that turn blue-green in color when stained with Giemsa. It is now recognized that the disorder may either be inherited or represent a secondary phenomenon because of an underlying systemic illness. SBHS is exceptionally rare with about 60 reported cases.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


