Junctional epidermolysis bullosa type Herlitz (JEB-H) is the autosomal recessively inherited, more severe variant of “lucidolytic” JEB. Characterized by generalized, extensive mucocutaneous blistering at birth and early lethality, this devastating condition is most often caused by homozygous null mutations in the genes LAMA3 , LAMB3 , or LAMC2 , each encoding for 1 of the 3 chains of the heterotrimer laminin-332. The JEB-H subtype usually presents as a severe and clinically diverse variant of the EB group of mechanobullous genodermatoses. This article outlines the epidemiology, presentation, and diagnosis of JEB-H. Morbidity and mortality are high, necessitating optimized protocols for early (including prenatal) diagnosis and palliative care. Gene therapy remains the most promising perspective.
Junctional epidermolysis bullosa type Herlitz (JEB-H) is the autosomal recessively inherited, more severe variant of “lucidolytic” JEB. Characterized by generalized, extensive mucocutaneous blistering at birth and early lethality, this devastating condition is most often caused by homozygous null mutations in the genes LAMA3 , LAMB3 , or LAMC2 , each encoding for 1 of the 3 chains of the heterotrimer laminin-332. The latter is a major adhesion protein within the basement membrane zone of the skin and mucous epithelia that provides stable anchorage of basal epithelial cells (keratinocytes) to the underlying dermis by connecting the hemidesmosomal component α6β4 integrin to collagen VII containing anchoring fibrils.
Epidemiology
Accuracy and comparability of epidemiologic data regarding the genodermatosis epidermolysis bullosa (EB), a rare “orphan” disease with complex phenotype-genotype correlations, are based on the limitation of bias by misdiagnosis, misclassification, restricted access to expert physicians with specific expertise, and differential enrolment in registries across the highly variable (clinical and genetic) spectrum of EB.
In an intention to overcome these obstacles, the United States National EB Registry (US NEBR) was founded in 1986 by the National Institutes of Health. This registry became the world’s largest cohort of well-characterized and monitored EB patients, and currently comprises more than 3200 patients whose demographics have been shown to closely mirror that of the entire American population, as well that of EB patient cohorts elsewhere in the world. Seven percent of these EB patients have some form of JEB, of which about 20% suffer from JEB-H. By extrapolation, prevalence and incidence rates of JEB-H have been estimated to be 0.07 and less than 0.41 per million, respectively.
In the following discussion on JEB-H, the authors refer to the most comprehensive and representative data provided by the US NEBR. When comparing this information with observations at the EB House Austria, an interdisciplinary clinical unit established in 2005 for state of the art medical care, academic affairs (education and training for laypersons and experts), diagnostics, and research, there is an overall corroboration/concurrence between the index populations with just one exception. In the authors’ experience, which is based on 6 genetically characterized JEB-H individuals currently documented in the Austrian EB Registry ( Table 1 ) and several international consultation cases, death in infancy or early childhood, despite the most aggressive therapeutic interventions, is the norm (ie, 100%). Moreover, registry data on JEB-H in general is rather believed to actually underreport mortality as infants with rapid demise may not have been referred for inclusion. This perception is in accordance with most other clinicians. The authors accordingly have never observed Herlitz patients surviving beyond the first year of life (mean lifetime 5.08 months), and thus none of the long-term skin or extracutaneous complications of JEB-H disease discussed in this article when referring to Herlitz NEBR patients. Those (even within the NEBR study population very rare) cases of long-term survival may either reflect a spectrum of disease severity mediated by genetic and epigenetic factors to be further characterized, or limited/restricted diagnostic validity/validation. For optimization of the latter, molecular mutation analysis in addition to structural analyses remains the current gold standard.
| Patient | Mutated Gene | Mutation | Survival Period (Months) |
|---|---|---|---|
| 1 | LAMB3 | R635X/R635X | 2 |
| 2 | LAMB3 | R635X/R635X | 8.5 |
| 3 | LAMB3 | R635X/R635X | 5.5 |
| 4 | LAMB3 | R635X/R635X | 6 |
| 5 | LAMC2 | L1122X/L1122X | 2.5 |
| 6 | LAMB3 | R635X/1629insG | 6 |
Clinical presentation of JEB-H
Skin
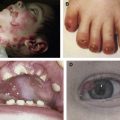
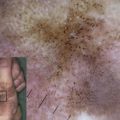
Reflecting profound mechanical fragility as the hallmark feature, at birth JEB-H patients present with extensive, generalized, recurrent and often persistent blistering, erosions, and crusting that cover not only particularly exposed skin areas (like palms and soles) but most or almost all of the body surface ( Fig. 1 ). Secondary lesions following chronic, repeated (even intrauterine) tissue traumatization include atrophic scarring, webbing (intradermal scar formation between fingers and toes), contractures (typically in axillary vaults), and milia (white papules).
Consecutive pigmentary abnormalities (hyper-, hypo, de-, or mottled pigmentation) rarely also comprise EB nevi (common in non-Herlitz JEB [JEB-nH]), ie, large eruptive, asymmetric, often irregularly pigmented, highly dynamic melanocytic lesions with sharply demarcated borders that frequently arise in areas of preceding blisters and may clinically mimic malignant melanoma (see the article by Lanschuetzer and colleagues elsewhere in this issue).
Pseudosyndactyly due to repeated blistering on hands and feet, initially presenting as partial interdigital fusion, webbing, or synechiae formation, and followed by complete fusion of all of the digits to a keratinaceous cocoonlike structure, frequently causes marked functional disability, muscle atrophy, and bone absorption. Exuberant granulation tissue, typically arising symmetrically around the mouth, central face, or nose as well as on the upper back, in axillary vaults, and around nail folds, is almost pathognomonic of JEB-H. Periorificial vegetation may cause complications such as total occlusion of nares, and implies therapeutic intervention by laser and sharp dissection. Differential diagnosis from squamous cell carcinoma is sometimes challenging, and may thus necessitate continuous clinical and occasionally histopathological evaluation. In addition, onychodystrophy with thickened, yellowish, longitudinally grooved, eventually marked curved and deformed nail plates (onychogryphosis), or absence (shedding) of nails due to atrophy and scarring of the nail bed (anonychia) are common findings in JEB-H.
Despite being more prominent in JEB-nH, localized or more diffuse scarring alopecia can also be observed in the Herlitz variant. Other uncommon cutaneous manifestations of JEB-H comprise palmoplantar keratoderma and congenital absence of skin with red, angulated, flame-shaped, well demarcated, depressed patches, usually unilateral, on hands, feet, wrists, or ankles.
Therapy in general is symptomatic, with antiseptic baths and functional bandages, splints, physical therapy, and surgical corrections (with often very high peri-interventional risk), thereby emphasizing the importance of preventing blistering by cool environment, avoidance of overheating, skin lubrication, water or air mattress, and soft, nonirritating fabric clothing.
Extracutaneous Involvement
Prototypical EB lesions such as blisters and erosions, followed by strictures, contractures, and stenoses, also occur in (epithelial) tissue outside the skin, involving for example the mucous membranes of the gastrointestinal, upper respiratory, and genitourinary tracts, the kidney, and external eye. These complex affections make JEB-H a systemic, multidimensional disease with a considerably high morbidity and mortality.
Ophthalmic findings in the Herlitz subtype include ocular surface (corneal, conjuctival) and eyelid abnormalities (blisters, erosions, and scarring), ectropion, exposure keratitis, pannus formation, limbal broadening, symblepharon, and lacrimal duct obstruction. Treatment is focused on ophthalmologic surgery and is accompanied by oral analgesics and topical antibiotics, as well as lubricants, gels, artificial tears, and bandages to reduce friction of the lids over the eye.
Upper respiratory tract involvement is a frequent phenomenon in both subtypes of JEB. The most common complications, namely chronic hoarseness, weak cry, or inspiratory stridor, are seen in up to 50% of all patients with JEB-H. End-stage sequelae are laryngeal webs, stenosis, and (acute) airway obstruction due to occlusion by blisters, diffuse edema, progressive scar formation, and strictures or exuberant granulation tissue following blistering on the edges of the laryngeal cords. The cumulative risk of acute airway obstruction in JEB-H determined by analyses of the NEBR population is about 13% by as early as age 1 year. In patients surviving beyond the neonatal period, it plateaus at a risk of nearly 40% by age 6 years and thereafter decreases again, most likely due to the age-related increase in luminal diameter of airways. Consequently, early and regular surveillance is mandatory. Means of therapeutic management include dexamethasone and adrenalin nebulizers, humidified oxygen, (early elective) tracheostomy, laser and sharp dissection, or topical mitomycin C to reduce granulation tissue.
Chronic otitis media is another symptom that is more common in JEB-H patients than in the general population. Chronic otitis media is again the result of a barrier-impaired cutis and a consecutively higher risk of microbial colonization and infection. Otological therapy includes local antisepsis and topical antibacterial agents, although resistance due to long-term use is a serious concern, especially in a group of patients prone to septic complications.
Intraoral disease is characterized by severe involvement of the oral soft and hard tissue, making both prophylactic and therapeutic approaches very challenging. Excessive caries results from a deficient oral hygiene due to painful peri- and intraoral blistering, erosions, and scar formation with consecutive contractures (microstomia, ankyloglossia), limited tongue mobility, and food clearance. Moreover, the specific, extremely cariogenic diet, a generally higher risk of infections such as candidiasis (reflecting the intrinsic barrier deficiency that stimulates colonization and invasiveness), as well as malnutrition (and consequent weakening of the immune system) are exacerbating factors that contribute to premature loss of teeth.
Enamel hypoplasia (pitting and furrowing of thin enamel, ultrastructurally displaying defects in prism structure and orientation) is another caries trigger and a characteristic feature of all JEB subtypes. Enamel hypoplasia is suggested to reflect a deregulated interaction of mutated adhesion proteins in the course of odontogenesis, histomorphogenesis, and cytodifferentiation. The spatially and temporally aberrant expression of mutated laminin-332 by enamel-forming ameloblasts may thus interfere with the intercellularly orchestrated regulation of enamel formation, signal transduction, cell polarity, mechanical stabilization and nutritional supply of enamel layers, enamel mineralization, orientation, and deposition at the enamel/dentin junction. The consequent disorganization and dysfunction of the basement membrane zone and the extracellular as well as enamel-forming matrix could represent the substrate of the abnormal dental architecture in JEB.
Development of highly aggressive squamous cell carcinomas was rarely reported in JEB-H patients surviving the early childhood period. Representing a dramatic long-term sequela arising most prominently in recessive dystrophic EB as early as within the second decade of life, ongoing nonhealing ulceration and consecutively permanent activation of reparative and proliferative pathways in combination with an intrinsically impaired intercellular and cell-matrix (structural as well as functional) regulation in EB are pathogenic sequences suspected to increase the risk of malignant transformation and development of tumors in the oral cavity (and skin).
Regular follow-ups by the dentist, emphasis on preventive measures (aggressive oral hygiene, fluoride substitution, reduction of cariogenic nutrition), and invasive approaches individually adopted to the patient’s general condition and prognosis, including usage of stainless steel crowns to minimize enamel destruction and maintain normal spacing, restoration of enamel and dentin defects with fillings to guarantee structure and continued function of teeth, lubrication of oral tissues to reduce shear forces, and extraction of most severely affected teeth with osteolytic foci to remove continuous sources of oral infections are strategies to ameliorate the oral status of affected individuals and to allow early recognition of malignancies.
Urologic abnormalities in EB occur with the highest frequency in patients with the Herlitz variant of JEB. Urethral meatus stenosis is the most common complication, observed in 11.6% of JEB-H patients within the NEBR, followed by urinary retention, hydronephrosis, and bladder hypertrophy in 9.3%, 7.0%, and 4.6%, respectively. Diverticuli within the urinary tract; scarring of the glans penis, hypospadias, epispadias; fusion of labia, narrowing of the vaginal vestibule, urinary reflux in vagina and uterine cavity; bladder-blistering, -edema, -cystitis, -infections, -extrophy, and reduced bladder capacity; ureteral stenosis, fibrosis, hydroureter; renal pelvis stenosis, pyelonephritis, recurrent urosepsis, and renal insufficiency are additional symptoms of JEB-H affecting the genitourinary tract. Dilatation, stent placement, resection, ureteral reimplantation, urethral catheterization, meatotomy, vesicostomy, or nephrostomy tube placement are some of the surgical interventions performed aiming at symptomatic or palliative relief.
The spectrum of gastrointestinal symptoms comprises dysphagia, esophageal stenosis or strictures, gastrointestinal reflux disease, peptic ulcer disease, and malabsorption. Constipation due to painful anal strictures and fissures, limited oral food (fiber) intake, and excessive loss of fluid through lesional skin is often exacerbated by opioid analgesia or sedatives, and may lead to life-threatening complications such as megacolon and perforation.
Together with recurrent generalized blistering and continuous transcutaneous loss of nutrients as well as a hypercatabolic state (reflecting an extraordinarily high energy consumption by wound healing, infections, and natural growth), chronic and severe gastrointestinal affection with protein-losing enteropathy can seriously compromise the nutritional status in JEB-H patients. An initially good weight gain is thus usually followed by a profound failure to thrive and, together with infectious/septic and respiratory complications, is associated with a high mortality in early childhood (see later discussion). Further effects of gastrointestinal affection by EB comprise a profound multifactorial anemia (chronic blood, iron, protein loss from open wounds and erosions within intestinal tract, poor intake and absorption of iron and other nutrients) or osteoporosis/osteopenia consequent not only to chronic malnutrition and malabsorption but also immobility/immobilization and concurrent renal insufficiency. Besides mediating growth retardation and dystrophy, nutritional deficiencies further accentuate an already intrinsically impaired wound healing.
Facing this broad spectrum of serious complications, global supplementation (including specialized formula feeds, calcium and vitamin D, oral bisphosphonate therapy) is usually required in JEB-H individuals. Moreover, esophageal dilatation or gastrostomy feeding are often indicated, although these techniques harbor a significant peri-interventional risk (eg, poor healing around the entry site) and a generally high rate of recurrence. Therefore, such procedures should be regarded as part of palliative care to improve comfort and quality of life.
Due to extensive disease, mortality (ie, death related to JEB-H) is very high, especially in the first year of life. Causes of premature death most commonly include recurrent infections and massive sepsis due to facilitated transcutaneous entry of Staphylococcus aureus , Streptococcus , Candida , or methicillin-resistant S. aureus via widespread skin erosions, pneumonia, respiratory failure other than pneumonia (eg, tracheolaryngeal obstruction), septic embolism, failure to thrive, and renal failure. Based on calculations assessing the NEBR population, the cumulative risk of death among JEB-H children by age 1 year is 44.7%.
As any medical intervention alleviates disease symptoms and complications at best but does not ultimately limit mortality, treatment approaches in Herlitz EB should be guided by ethical principles and norms focusing on life comfort and company (see Yan and colleagues for an excellent review).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


