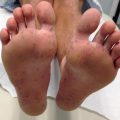
Fig. 6.1
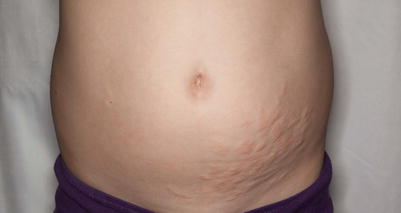
Ehlers-Danlos syndrome. Increased skin laxity
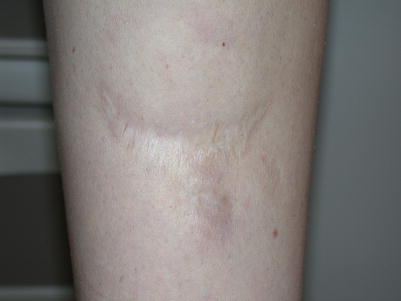
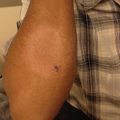
Fig. 6.2
Ehlers-Danlos syndrome. Widened fish-mouth scar with atrophic skin on the leg
The clinical features of vascular EDS can be very subtle, and at times the diagnosis is only made after a life-threatening vessel rupture or sudden death in the third or fourth decade of life. The presenting feature in childhood is often extensive, unexplainable bruising at sites not prone to trauma. Unlike classical EDS, the skin in vascular EDS is not hyperextensible, though does tend to be thin, with visible underlying vessels, particularly on the trunk. Some patients have a characteristic facies with a thin, pinched nose, prominent eyes, lobeless ears, and a lack of subcutaneous fat. Joint hypermobility is generally restricted to the small joints of the hands. The most concerning features of vascular EDS are arterial dissection, rupture, and aneurysm. Bowel and uterine rupture may also occur. Pneumothorax may be a more common manifestation in childhood, as vessel and hollow organ rupture typically do not occur until the third or fourth decades of life. Vascular EDS is caused by mutations in type III collagen.
Hypermobile EDS remains a diagnostic challenge, as there is no diagnostic test or established genetic basis to aid in diagnosis. In addition, joint hypermobility can be dominantly inherited in many families, and it continues to be debated whether hypermobile EDS and joint hypermobility syndrome actually represent the same condition. There are no life-threatening complications associated with this form of EDS, though patients can have significant morbidity related to chronic joint pain. Patients are also more likely to develop postural orthostatic tachycardia syndrome (POTS) and other forms of autonomic dysfunction that can negatively impact quality of life.
Management Strategies
For all suspected EDS patients, a full history and physical exam are necessary, including a detailed pedigree. Clinical examination should include a full skin exam, as well as assessment of the joints. Skin biopsy may be helpful in patients where certain subtypes are being considered.
Patients with classical EDS have a wide variation in clinical severity. All patients should follow regularly with cardiology, with joint hypermobility managed by rheumatology, physical therapy, and occupational therapy. Patients should make any surgeons aware of their condition to allow for appropriate suturing techniques.
For vascular EDS, in addition to the standard history and physical exam, baseline imaging of the arterial tree should be included. This can be accomplished with computed tomography angiography (CTA) at a young age to minimize sedation, but eventually should transition to magnetic resonance angiography (MRA). Conservative medical management is preferred when possible, as overly aggressive surgical and endovascular interventions can cause unnecessary morbidity and mortality. Patients should follow regularly with cardiology, though the role for repeat imaging and preventative medication is not well defined at this time. A recent study with a beta-blocker celeprilol found a significant decrease in arterial events, suggesting some benefit in complication prevention in these patients. Angiotensin receptor blockers have shown additional promise in delaying complications in animal models, and may play an increasing role in management of these patients in the future. For self-management, patients with vascular EDS should avoid potentially harmful activities, such as contact sports, heavy lifting, and any cause of rapid acceleration and deceleration.
Patients with hypermobile EDS should be followed by rheumatology, physiotherapy, and occupational therapy. Referral to pain clinics can be helpful to manage the significant pain that can develop over time with this condition. Connection with support groups as well as proper counseling by a psychologist or a psychiatrist can be beneficial to affected patients. The Ehlers Danlos National Foundation (EDNF) provides educational resources for patients to learn about their diseases and connect with each other.
Specific Investigations Recommended [4]
For diagnosis |
Skin examination |
Joint examination for hypermobility |
Skin biopsies for histology, electron microscopy, and fibroblast culture |
Genetic testing |
Cardiology assessment including echocardiogram |
Carotid duplex ultrasound (vascular EDS) |
Full body MR angiography (vascular EDS) |
For monitoring |
Regular cardiology follow-up |
Annual carotid and abdominal ultrasounds for arterial screening in asymptomatic patients (vascular EDS) |
Serial MR angiography every 6–12 months with known arterial complications (vascular EDS) |
DEXA scan at diagnosis especially in those at risk |
First Line Therapies
As discussed above, most management of patients with EDS is directed toward appropriate monitoring and referrals for supportive care. Patients with EDS are predisposed to functional bowel disorders (i.e. gastritis and irritable bowel disease), low blood pressure, pain, temporomandibular disorder, and gum disease. Proper management of these disease manifestations by specialists is of important therapeutic consideration. Multidisciplinary clinics around the country are forming to improve management of these patients. There is limited medical treatment available currently to prevent symptoms associated with vascular EDS. Beta-blockers and angiotensin receptor blockers show promise as possible preventative measures in patients with vascular EDS.
Celiprolol 100–400 mg twice daily (vascular EDS) | A |
Losartan 2 mg/kg/day (vascular EDS) |
Marfan Syndrome
Clinical Features
Marfan Syndrome is an autosomal dominant inherited disorder of connective tissue caused by mutations in fibrillin 1, an important structural component of the microfibrils which compose elastin and other connective tissues, with a highly variable clinical presentation. The diagnosis is made clinically based on family history, mutational analysis, and hallmark criteria involving three organ systems: the ocular, skeletal, and cardiovascular systems. The National Marfan Foundation Website contains the most current diagnostic criteria necessary for a diagnosis.
Ectopia lentis or displacement of the lens from the center of the pupil affects 60 % of patients with Marfan Syndrome and is an important diagnostic feature. Other visual findings include myopia (the most common visual finding), retinal detachment, glaucoma, and early cataract formation.
Joint laxity and bone overgrowth are also characteristic findings, and the changes in the skeleton are progressive during periods of rapid growth during childhood. Overgrowth of the bones leads to extremities which are disproportionately long for the size of the body known as dolichostenomelia. Rib bone overgrowth may lead to pectus excavatum or pectus carinatum. Scoliosis is common and may become progressively worse. Assessment for the skeletal features characteristic of Marfan Syndrome is important when calculating a patient’s Systemic Score, a clinical assessment tool used when establishing a diagnosis of Marfan syndrome, also available at the National Marfan Foundation Website. The different skeletal features that are considered and weighted include the following: pectus carinatum, pectus excavatum, chest asymmetry, hindfoot deformity, pes planus, scoliosis, thoracolumbar kyphosis, reduced elbow extension, a reduced ratio of the upper body segment length to the lower body segment length, an increased arm span to height ratio, and protrusio acetabulae which refers to an abnormally deep acetabulum with accelerated erosion. The thumb sign, when the thumb is noted to extend beyond the palm when the patient makes a fist, and the wrist sign, when the thumb and fifth digit are able to overlap when wrapped around the patient’s other wrist, are also skeletal findings that increase a patient’s Systemic Score and make a diagnosis more likely. Characteristic skeletal facial features to consider include a long narrow face, enophthalmos, downward slanting palpebral fissures, malar hypoplasia, micrognathia, and a highly arched palate with crowding of teeth.
The most serious manifestations of Marfan Syndrome affect the cardiovascular system and include progressive aortic root dilatation with a risk of aortic dissection or rupture, typically in adulthood, as well as aortic insufficiency. Other cardiac findings include mitral valve prolapse with or without mitral regurgitation and tricuspid valve prolapse with possible enlargement of the proximal pulmonary artery. Although mitral valve prolapse and mitral regurgitation is typically tolerable for the majority of patients, the leading cause of cardiovascular morbidity and mortality and the main indication for cardiovascular surgery in children with severe Marfan syndrome is mitral valve prolapse with associated ventricular dysfunction. Marfan syndrome patients are at an especially increased risk of cardiac complications while pregnant due to risk of rapid aortic root enlargement and aortic dissection or rupture during pregnancy, delivery and the postpartum period. A sufficiently dilated aortic root, calculated as a Z score which takes into account norms for patient’s age and size, is used as a diagnostic criteria for Marfan syndrome while mitral valve prolapse is factored into the Systemic Score.
The only cutaneous finding in Marfan Syndrome is striae distensae. Other diagnostic findings include frequent hernias, lung bullae which may progress into spontaneous pneumothorax, and dural ectasia or stretching of the dural sac in the lumbosacral region which puts patient at risk for bone erosion, nerve entrapment, and CSF leaks. The presence of any of these findings adds to the Systemic Score.
Management Strategies
Management of Marfan Sydrome is aimed at diagnosing the condition, trying to prevent cardiovascular complications, and managing associated complications. Evaluations by specialists in ophthalmology, orthopedics, cardiology, and medical genetics who are familiar with Marfan Syndrome are necessary to establish the diagnosis, and determine the extent of disease. Management depends on severity of involvement but typically involves a multidisciplinary approach using genetics, cardiology, ophthalmology, orthopedics, and cardiovascular surgery.
The highest morbidity and mortality in affected patients is related to cardiovascular disease but the life expectancy approximates that of the general population with proper management of the cardiovascular manifestations. Medication management with agents that reduce hemodynamic stress on the aorta are paramount with progression to cardiothoracic surgical intervention when the aorta becomes excessively dilated or when ventricular dysfunction develops from the mitral regurgitation.
Specific Investigations Recommended
For diagnosis |
• Detailed medical history |
• Detailed family pedigree |
• Complete examination with special attention to skeletal abnormalities and striae distensae by medical genetics or physician with expertise in Marfan Syndrome |
• Ophthalmologic exam including slit lamp exam looking for lens subluxation |
• Echocardiogram to measure aortic root and assess cardiac valves |
• Genetic testing for fibrillin 1 mutation |
For treatment |
• Regular evaluation by medical geneticist |
• Annual ophthalmologic evaluation to assess vision acuity and the development of glaucoma and/or cataracts |
• Evaluation by an orthopedist to manage scoliosis and other skeletal abnormalities |
• Regular evaluation by a cardiologist; frequency of visits determined by severity of disease |
• Serial echocardiogram annually to monitor aorta and cardiac valve; more frequently when aortic root diameter > 4.5 cm or when the rate of aortic dilation > 0.5 cm per year |
• Pulmonary imaging with CXR or high resolution CT when concern for pneumothorax |
• MRI of lumbosacral spine when concern for dural ectasia |
• Care by a high risk obstetrician with pregnancy |
First Line Therapies
Beta blocker therapy should be initiated in all patients at time of diagnosis and continued throughout their lifetime to reduce hemodynamic stress on the aorta wall and to prevent dilatation based upon recommendations of the American Heart Association and American College of Cardiology. Atenolol is generally the preferred medication due to selectivity for beta 1 receptors although propranolol has been used as well. – A [5–7]
Aggressive early correction of vision abnormalities with glasses to prevent amblyopia – E
Antibiotics for prevention of subacute bacterial endocarditis prior to dental procedures in patients with valve abnormalities [8]
Orthotics for symptomatic pes planus – E
Palate expander and orthodontia when indicated – E
Bracing for scoliosis when indicated – E
Second Line Therapies
Losartan, an ACE-inhibitor, has recently been found in a randomized trial to be superior to placebo in preventing aotic root dilatation over 3 years of use. Another study comparing losartan to atenolol found a similar rate of reduction of aortic root dilatation with both medicines. Questions remain as to appropriate dosing of losartan for this indication, timing of initiation of therapy, and whether losartan and beta blockers should be used individually or together – A [9, 10]
Hormone supplementation to accelerate puberty and limit adult height in cases where extreme adult height is predicted – E [11]
Surgical Considerations
Surgical repair of the aorta when the maximal measurement approaches 5.0 cm in adults or older children, the rate of increase of diameter nears 1.0 cm per year, or there is progressive and severe aortic regurgitation – D
Removal of the lens (surgical aphakia) when the lens is dislocated and mobile or interfering with vision – D
Surgical stabilization of severe scoliosis – D
Surgical correction of severe pectus excavatum – E
Surgical correction of symptomatic protusio acetabulae – E
Surgical correction of severe pes planus refractory to orthotics – E
Pleurodesis or removal of surgical blebs if recurrent pneumothorax – E
Use of supporting mesh when hernias are surgically repaired to prevent recurrence – E
Cutis Laxa
Clinical Features
Cutis laxa (CL) is a heterogenous group of inherited and acquired disorders characterized by abnormal elastic tissue in the skin and other organs [12]. The skin of cutis laxa tends to be hypoelastic, redundant, and hangs in folds most readily apparent in the axillae, groin, and hands (Fig. 6.3). Involvement of the face leads to a prematurely aged appearance. As opposed to the skin of Ehlers Danlos syndrome which stretches and recoils quickly, the skin of cutis laxa pulls away easily from underlying structures but does not return to its usual position quickly when released. It also does not bruise easily and is not characterized by poor scarring.
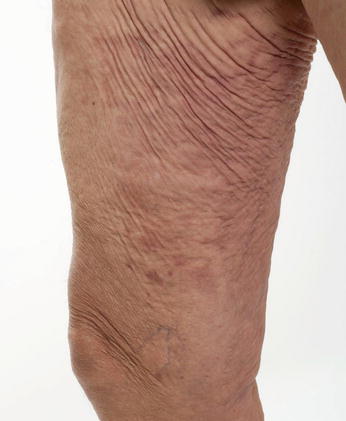
Fig. 6.3
Cutis laxa. Hypoelastic, redundant skin folds over the upper thigh and groin
Acquired CL typically occurs in adulthood and can follow medication use, an inflammatory process of the skin, or be associated with malignancy, autoimmune, or other systemic disease. We will focus here instead on the hereditary forms of cutis laxa, which are distinguishable based on their inheritance pattern, pathogenic mutation, and variable extracutaneous features, and present in infancy or childhood.
Autosomal dominant CL is due to a mutation in ELN resulting in a defective tropoelastin protein that is unable to bind properly to the fibrillin scaffold. It is highly variable even within families but characterized predominantly by cutis laxa of the skin that develops in childhood or early adulthood and progresses with time. Distinctive facial features include a long philtrum, high forehead, prominent ears with large lobes, and beaked nose. Patients have an increased tendancy to develop hernias. Cardiopulmonary manifestations range from mild to severe, and although many patients have a normal life span, some will develop bronchiectasis, emphysema, aortic aneurysms, and pulmonary artery disease.
The skin and ear findings of autosomal recessive cutis laxa (ARCL) type 1 are similar to those of the dominant form but are notable at birth. Other features include hernias, growth and motor delays, and a higher likelihood of systemic complications including emphysema, diaphragmatic defect, tortuous arteries, and aneurysms leading to a high risk of death from cardiopulmonary complications in childhood. Two variants of pathogenic mutations have been associated with ARCL which differ in their cardiovascular manifestations. Patients with FBLN4/EFEMP2 mutaions, also known as ARCL Type Ia are at risk of aortic aneurysm but not supravalvular aortic stenosis while those with FBLN5 mutations, also known as ARCL Type Ib, are at risk of supravalvular aortic stenosis but not aneurysm. In addition, ARCL Type Ia has characteristic retrognathia, hypertelorism, joint laxity and congenital hip dislocation, while ARCL type Ib lacks these features and has bladder diverticulae.
Autosomal recessive CL type II comes in two variants distinguished by their pathogenic mutations and clinical features. ARCL-IIA is caused by a mutation in ATP6VOA2, which leads to problems with secretion of tropoelastin, while ARCL-IIB is caused by a mutation in PYCR1 and a resulting defect in proline metabolism. While both share common features, including inelasticity of skin, retrognathia, hernias, mental retardation, intrauterine growth retardation, postnatal growth delay, and scoliosis, ARCL-IIA specifically is associated with aortic aneurysm, bladder diverticula, patent anterior fontanelle, hypotonia, and delayed motor development. On the other hand, ARCL-IIB has unique features of osteoporosis, athetoid movements, and corneal opacities.
A less known severe variant of cutis laxa is autosomal recessive CL type III (Progeroid syndrome of De Barsy or CL-corneal clouding-mental retardation syndrome) characterized by progeroid appearance, reduced subcutaneous fat, corneal opacities, athetoid movements, and mental retardation. Another rare variant known as autosomal recessive CL with severe pulmonary, gastrointestinal and urinary abnormalities (Urban-Rifkin-Davis syndrome) is distinguished by emphysema, atelectasis, tracheomalacia, diaphragmatic hernia, hydronephrosis, and diverticulae of the bladder and gastrointestinal tract, which is frequently fatal in infancy.
X-linked cutis laxa (XLCL), or Occipital Horn yyndrome, was formerly classified as a type of Ehlers-Danlos yyndrome. Allelic to Menkes disease, this variant of CL is caused by a defect in a copper transporting adenosine triphosphatase which leads to a functional deficiency of copper and impairs enzymes essential in elastic tissue production. As opposed to patients with Menkes who frequently die before age 4 due to severe neurologic defects, patients with XLCL have predominantly connective tissue abnormalities, including the hallmark downward-pointing exostoses on the occipital bone as well as inelastic skin with a droopy face at birth, occasional pili torti of hair, bladder diverticulae, urinary tract infections, inguinal hernias, and orthostatic hypotension.
Management Strategies
There is no cure for cutis laxa, and management is focused instead on making the diagnosis and then trying to prevent and manage associated complications. Once the diagnosis is suspected, affected patients should undergo comprehensive evaluation to identify the type of cutis laxa and extent of the condition and any associated systemic manifestations. Management of the patient involves a multidisciplinary approach, which varies depending on the type and extent of disease, but may involve specialists in genetics, plastic surgery, cardiology, neurology, cardiothoracic surgery, ophthalmology, gastroenterology, and urology. Because the extracutaneous manifestations differ substantially, the individual management of a patient must be specifically tailored and the recommendations listed below may not be clinically indicated or entirely complete for every patient.
For diagnosis |
• Skin biopsy with elastic stains to evaluate decreased elaunin fibers and fragmentation of elastic fibers in reticular dermis. Findings may be minimal and does not allow differentiation of subtype of cutis laxa |
• Skin biopsy for electron microscopy may further elucidate abnormalities in elastic tissue production and may assist in differentiation of cutis laxa type but not widely available |
• Referral to medical genetics for complete evaluation |
• Genetic testing for known pathogenic mutations |
• CXR |
• 3-dimensional CT scan |
• Pulmonary function tests |
• Echocardiogram |
• MRA from head to pelvis |
• Kidney ultrasound |
• Barium enema |
• Voiding cystouretogram |
For treatment |
• Serial CXR, CT scan, pulmonary function tests, echocardiogram, MRA, kidney ultrasound, barium enema, and voiding cystoureterogram as clinically indicated |
• Subspecialty referrals that are frequently needed: plastic surgery, pulmonary, general surgery, cardiology, urology, cardiothoracic surery, physical therapy |
First Line Therapies
Education on tobacco avoidance and sun protective measures – E
Plastic surgery correction of redundant skin folds – E
Symptomatic management of pulmonary emphysema with special attention prior to surgeries – E
Serial monitoring of aortic aneurysms and other arterial abnormalities with referral for surgical management as needed – E
Surgical management of hernias – E
Surgical management of diverticula – E
Pseudoxanthoma Elasticum
Clinical Features
Pseudoxanthoma elasticum (PXE) is a multisystem disorder of ectopic mineralization with cutaneous, ocular, and cardiovascular manifestations. The prevalence is around 1 in 50,000, with a slight female predominance. PXE is inherited in an autosomal recessive fashion and is caused by mutations in ABCC6, an ABC-cassette transporter located predominantly in hepatocytes, although its exact function remains unclear. Progressive calcification of elastic fibers in target organs occurs, leading to findings within the dermis of the skin, mid-sized arteries throughout the body, and Bruch’s membrane of the eye.
PXE most commonly presents with cutaneous findings, often in the first or second decade of life. Soft yellowish papules appear at the neck and other flexural areas (Fig. 6.4), with later coalescence into firmer plaques, along with the development of loose, redundant skin at the axillae and groin. Yellowish papules may also appear on mucosal surfaces, especially the lower lip, and prominent creases may be seen on the chin. Skin biopsy will reveal distorted, fragmented elastic fibers in the mid to deep reticular dermis, or in more advanced cases, calcium deposits on the elastic fibers.
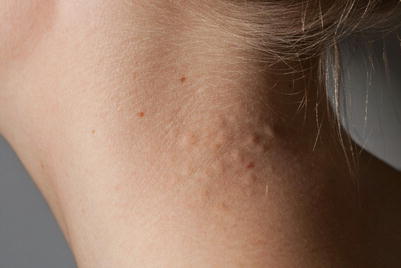
Fig. 6.4
Pseudoxanthoma elasticum. Soft, yellowish papules containing distorted fragmented elastic fibers on the neck
Angioid streaks of the eyes develop in nearly all PXE patients by the third decade of life. This finding is caused by breaks in the calcified elastic lamina of Bruch’s membrane, the innermost layer of choroid. Over time, this can lead to choroidal neovascularization, followed by hemorrhage and scarring, with progressive loss of vision and even blindness. Other ocular findings may be present, including mottled, “peau d’orange” changes of the retinal pigmented epithelium.
Cardiovascular manifestations are the primary cause of premature mortality from PXE. The consequences of arterial calcifications can include intermittent claudication, loss of peripheral pulses, renovascular hypertension, angina pectoris, myocardial infarction, and stroke. Within the gastrointestinal tract, arterial calcifications can lead to gastrointestinal hemorrhage.
Management Strategies
The diagnosis of PXE requires, at a minimum, the presence of retinal angioid streaks in combination with characteristic skin lesions that show the diagnostic histopathological findings of calcified dystrophic elastic fibers. The diagnosis may be also be made via molecular genetic testing for ABCC6 mutations, especially in patients who are too young to demonstrate the ocular or cutaneous manifestations.
Unfortunately, there currently is no proven treatment for the systemic mineralization seen in PXE. For all patients, a healthy lifestyle with diet, exercise, weight control, and avoidance of smoking and excessive alcohol should be emphasized in order to minimize other cardiovascular risk factors. Skin findings may be ameliorated by surgical excision of areas of redundant skin, or by the application of resurfacing lasers to improve the skin texture. Research on PXE has focused on elucidating the function of the ABCC6 transporter. It remains unclear what molecules are transported by the ABCC6 transporter. Recent research has shown that ABCC6 transporters in the liver mediate the cellular release of ATP, which is converted extracellularly into AMP and inorganic pyrophosphate (PPi), a mineralization inhibitor. Patients with PXE demonstrated lower plasma concentrations of PPi, suggesting that supplementation with PPi could represent a future avenue for treatment [15–18].
Studies in mouse models have shown that increasing the magnesium content fivefold that of a normal diet can prevent ectopic mineralization and, likewise, studies with magnesium-containing phosphate binders have shown similar effects. A clinical trial is currently under way to test the efficacy of supplemental dietary magnesium in human patients with PXE [19–27].
While studies in animal models have demonstrated accelerated mineralization in the setting of warfarin therapy, studies evaluating the role of supplemental vitamin K in preventing or slowing the mineralization process have been disappointing. Thus while warfarin therapy should be avoided if possible in PXE patients, supplemental vitamin K is not recommended [28–31].
In the past several years, case reports and series from the ophthalmology literature have documented the role of intravitreous injection of vascular endothelial growth factor receptor antagonists as a means to treat choroidal neovascularization and prevent loss of visual acuity and blindness. However, it should be noted that acute thromboembolic events have occurred following the administration of these medications [32–36]. Other treatments for choroidal neovascularization in patients with PXE have been attempted, including laser photocoagulation, transpupillary thermotherapy, macular translocation surgery, and photodynamic therapy, with predominantly disappointing results [37, 38].
Other areas of research include transplantation with hepatoblastic lineage cells or other pluripotent stem cells, as well as use of read-through molecules, which allow transcription of the ABCC6 gene even in the presence of nonsense mutations. These treatments have not been studied in humans, however [39, 40].
Investigations Recommended
For diagnosis |
Skin biopsy with stains for calcium (von Kossa) and elastin (Verhoeff-Van Gieson) |
Genetic testing for ABCC6 mutations for diagnosis or family planning |
Complete examination by ophthalmology |
For surveillance |
Biannual evaluation by ophthalmology |
Serial evaluation by cardiology |
Baseline EKG |
Baseline Echocardiogram |
Baseline and serial serum lipids |
Monitoring for black, tarry stools and referral to gastroenterology if necessary |
Referral to other specialties such as dermatology, vascular surgery, plastic surgery, or nutrition depending on patient presentation |
First Line Therapies
Fractional carbon dioxide laser for improvement in skin texture – E [43]
Intravitreal vascular endothelial growth factor receptor antagonists (bevacizumab or ranibizumab) for patients with choroidal neovascularization – D [32–36]
Other treatments should be tailored in consultation with specialists depending on disease presentation and severity
Elastosis Perforans Serpiginosa
Clinical Features
Elastosis perforans serpiginosa (EPS) is a rare form of perforating disorder that may be inherited or acquired. About 40 % of cases occur in conjunction with other genetic syndromes, including Down syndrome, scleroderma, Rothmund-Thomson syndrome, acrogeria, Marfan syndrome, osteogenesis imperfect, pseudoxanthoma elasticum, and Ehlers-Danlos syndrome. The medication penicillamine has also been reported to induce EPS. EPS can also be an isolated, idiopathic skin finding. Familial cases have been reported, with no consistent pattern of inheritance.
EPS presents with keratotic papules <5 mm arranged in an annular or serpiginous fashion. The lateral neck is the most common location, but lesions may occur on the upper extremities, face, or other flexural areas (Fig. 6.5). Most lesions are asymptomatic, but mild pruritus can occur. EPS often resolves spontaneously after a period of up to several years, but may leave atrophic scars. Biopsy of EPS shows transepidermal elimination of elastic fibers and inflammatory debris with overlying hyperkeratosis and surrounding epithelial hyperplasia. Verhoeff-van Gieson stain will help to identify elastic fibers both within the hyperkeratotic plug and the surrounding epidermis and superficial dermis.
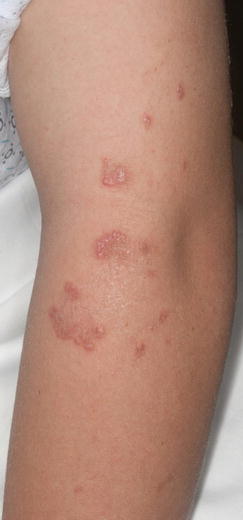
Fig. 6.5
Elastosis perforans serpiginosa. Grouped hyperkeratotic papules distributed in an annular or serpiginous fashion on the left upper arm
Management Strategies
When the diagnosis is uncertain clinically, a skin biopsy can be performed to help clarify the diagnosis [44]. A complete history and physical examination is warranted in all patients with EPS. Further testing for associated connective tissue disorders or genetic disorders should be guided by the results of the history and physical findings. Most clinicians do not routinely perform any further investigations in otherwise healthy patients.
In cases related to penicillamine therapy, the medication should be discontinued, although some case reports suggest that in susceptible patients, the damage is irreversible. There are many therapies that have been used anecdotally for EPS, but to date there have been no large studies or clinical trials, and there is no gold standard for treatment. All modalities of treatment have been associated with mixed success. In asymptomatic cases, observation may be appropriate, as the lesions do resolve spontaneously.
Specific Investigations Recommended
For diagnosis |
Skin biopsy |
Thorough history and physical examination |
First Line Therapies
Second Line Therapies
Intralesional steroid (triamcinolone acetonide 40 mg/ml every 15 days for 3 months) plus topical allium cepa-allantoin-pentaglycan gel (twice daily) – E [57]
Aminolevulinic acid (7.6 % topical solution) with photodynamic therapy – E [61]
Cellophane tape stripping – E [45]
Narrow band ultraviolet B phototherapy for disseminated lesions – E [45]
Local excision – E [45]
Dermabrasion – E [45]
Focal Dermal Hypoplasia
Clinical Features
Focal dermal hypoplasia (FDH) is a rare, X-linked dominant condition (95 % of cases are sporadic) that primarily affects female infants, although it may be seen in males who are mosaic for mutations in the PORCN gene. Clinical features of FDH are seen at birth and may affect multiple organ systems, most commonly the skin, skeletal system, and eyes.
Cutaneous findings may occur anywhere on the skin, and consist of atrophic linear erythematous areas of skin and/or hypo- or hyperpigmentation that often follows the lines of Blaschko (Fig. 6.6). Large areas of aplasia that subsequentlyheal with atrophy may also be present, accompanied by soft, yellow to brown outpouchings that are caused by herniation of fat through the dermis (Fig. 6.7). Patients commonly go on to develop erythematous papillomas at mucocutaneous junctions that can erode, ulcerate, and bleed. Hair may be sparse or absent on the scalp or other areas of the body, and nails may be dysplastic or hypoplastic. Approximately 80 % of patients will also have skeletal abnormalities. These are varied but can include syndactyly, hypoplasia or absence of digits, polydactyly, scoliosis, costovertebral abnormalities, facial asymmetry, or the rare but unique “lobster claw” deformity. Osteopathic striae may be seen radiologically and may be a helpful clue when the diagnosis is uncertain.
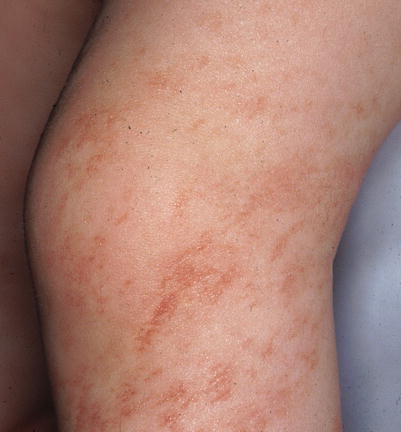
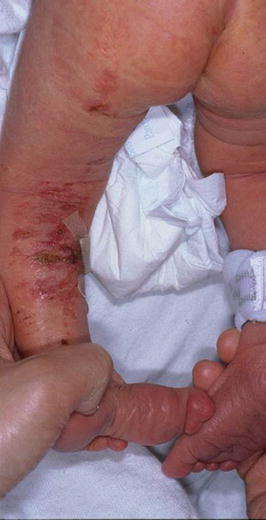
Fig. 6.6
Focal dermal hypoplasia. Linear atrophic erythematous patches on the leg follows the lines of Blaschko
Fig. 6.7
Focal dermal hypoplasia. Large areas of linear patches of aplasia that subsequently healed with atrophy. These areas may also be seen with soft, yellow to brown outpouchings that are caused by herniation of fat through the dermis
Eye abnormalities are seen in approximately 40 % of patients. They are usually present at birth, and can range in severity from having no impact on vision to blindness. Ocular abnormalities may include colobomas, strabismus, nystagmus, anophthalmia, microphthalmia, aniridia, heterochromia, cataracts, and hypopigmented/hyperpigmented retina. Other organ systems that may be affected include the dental, gastrointestinal, and urogenital systems.
Management Strategies
Management of FDH initially focuses on making an accurate diagnosis, and later on treating manifestations of the disease.
There is no cure for FDH, and treatment is largely supportive. For skin manifestations, care is directed at treating open wounds, preventing infection, and treating excessive granulation tissue. Excessive granulation tissue impedes wound healing in many patients. For those with skeletal manifestations, physical/occupational therapy and surgical intervention may be beneficial, but recommendations are specific to the type of manifestation and complications specific to the patient. Consultations with multiple subspecialists may be required as follows.
Specific Investigations Recommended
Skin biopsy of affected skin reveals a normal epidermis that overlies a hypoplastic dermis with reduced collagen. Islands of fat cells are present within the superficial dermis, clinically consistent with herniation of fatty tissue.
Eye examination to evaluate for ocular abnormalities
Hearing evaluation to evaluate for hearing defects
Chest X-ray to evaluate for costovertebral defects or diaphragmatic hernia
Renal ultrasound to evaluate for structural genitourinary abnormalities
Genetic testing may be considered to look for PORCN gene mutations
For diagnosis |
Skin biopsy |
Eye examination |
Hearing evaluation |
Chest X-ray |
Renal ultrasound |
Genetic testing |
For surveillance |
Orthopedic surgery- if skeletal abnormalities present; regular exams to evaluate for scoliosis |
Dental specialties- regular exams are recommended |
Ophthalmology- baseline evaluation and continued follow-up if abnormalities present |
Pediatric surgery- if diaphragmatic hernia or abdominal wall defects present |
Urology- if abnormalities present |
First Line Therapies
Wound care and topical antibiotics: for those with significant erosions and areas of aplasia, wound care with topical antibiotics to prevent secondary infection, and use of occlusive dressings until epidermal healing occurs is recommended [64].
Physical/occupational therapy is recommended for those with impaired function due to their skeletal abnormalities [64].
Topical antibiotics | E |
Wound care | E |
Physical/occupational therapy | E |
Second Line Therapies
Excessive granulation tissue can be very problematic for patients with FDH. Each of the following therapies are generally regarded as conventional treatments for excessive granulation tissue in patients with FDH. (Evidence level E) [64–66]
Silver nitrate
Topical or intralesional steroids
Cryotherapy/curettage
Surgical excision
Third Line Therapies
Pulsed dye laser- treatment of the cutaneous lesions of FDH can be difficult to treat. One report described successful decrease in the erythema associated with one patient’s atrophic lesions of FDH (Evidence level E) [65]. Photodynamic therapy: Excessive granulation tissue in FDH can be very refractory to conventional means of treatment. Two patients – one adult, and one child – have had successful treatment of excessive granulation tissue refractory to conventional modalities with photodynamic therapy (Evidence level E) [66].
Buschke-Ollendorff Syndrome
Clinical Features
Buschke-Ollendorff syndrome is a rare autosomal dominant genetic connective tissue disorder that is caused by loss-of-function mutations in LEMD3 and is characterized by the presence of connective tissue nevi and osteopoikilosis. Connective tissue nevi generally present in early childhood, but may be present at birth. While the connective tissue nevi can be composed of either elastin or collagen, most are elastomas. When composed of collagen, findings are consistent with dermatofibrosis lenticularis disseminate. They connective tissue nevi are typically asymptomatic, firm flesh-colored to yellow papules and nodules that may coalesce into plaques (Fig. 6.8). They are most commonly asymmetrically present on the trunk or extremities, but may also be present in other areas of the skin, such as the skin folds. They typically grow in proportion to the child, and the number of lesions present may increase with age.