Hereditary Disorders of Genome Instability and DNA Repair: Introduction
|
Because the genome exerts control for cellular function, maintaining genome stability is important for the continued function of cells, tissues, and organisms. DNA is the carrier of genetic information. Its structure is regularly threatened by damaging agents that include oxidative stress, ultraviolet (UV) and X-radiation, and chemical agents. Although much damage is repaired, failure to maintain genomic integrity may lead to abnormal cell function or cell death. If the cell divides, progeny may accumulate additional damage and this progressive accumulation of damage can lead to malignancy (see Chapter 110).
This chapter describes the relevant skin disorders with genome instability and the underlying defective mechanisms of DNA repair or DNA maintenance (Tables 139-1 and 139-2). All of these exhibit prominent cutaneous abnormalities that involve dermatologists in their diagnosis and management. Most, but not all, are also characterized by an increased risk of malignancies. This demonstrates that the maintenance of genome integrity is of utmost importance for the prevention of malignant transformation. Malignant transformation requires the accumulation of several mutations in specific genes of a single cell. Thus a mutator phenotype is often regarded a prerequisite for carcinogenesis, because without genome instability it would be exceedingly unlikely that all of those mutations occur in a single cell.1–4 The same genes that are affected in the hereditary genome instability disorders can also confer genome instability to individual cells when impaired through acquired mutations, thereby playing an important role early in spontaneous carcinogenesis.
Disorder | Clinical Features | |||
|---|---|---|---|---|
Cutaneous | Neoplasia | Other | Inheritance | |
Disorders of Genome Instability with defective DNA repair | ||||
Xeroderma pigmentosum (XP) | Photosensitivity (blistering on minimal sun exposure in some patients), freckle-like (lentiginous) macules, poikiloderma (hyper- and hypopigmentation, atrophy, telangiectasia), skin cancer | BCC, SCC, melanoma, central nervous system tumors | Sensorineural deafness, progressive neurologic degeneration, primary loss of neurons (some patients) | Autosomal recessive |
Cockayne syndrome (CS) | Photosensitivity (burning on sun exposure) | No increased incidence | Typical facial features (deep set eyes, loss of subcutaneous fat), pigmentary retinal degeneration, postnatal growth failure, sensorineural deafness, progressive neurologic degeneration, primary dysmyelination, brain calcifications | Autosomal recessive |
Xeroderma pigmentosum/Cockayne syndrome complex (XP/CS) | Photosensitivity (burning on sun exposure), freckle-like (lentiginous) macules, poikiloderma (hyper- and hypopigmentation, atrophy, telangiectasia), skin cancer | BCC, SCC | Neurologic changes of CS | Autosomal recessive |
Trichothiodystrophy’ (TTD) | Brittle hair, photosensitivity (burning on sun exposure), ichthyosis, collodion membrane, “tiger tail banding” of hair with polarized microscopy | No increased incidence | Congenital cataracts, short stature developmental delay, microcephaly, primary dysmyelination, recurrent infections | Autosomal recessive |
Xeroderma pigmentosum/trichothiodystrophy (XP/TTD) | Clinical features of both TTD and XP including skin cancer | BCC, SCC, melanoma | Clinical features of both TTD and XP | Autosomal recessive |
Hereditary nonpolyposis colon cancer (HNPCC)/MUIR–TORRE Syndrome (MTS) | Sebaceous tumors (benign and malignant), keratoacanthomas | Low-grade cancer of the colon, endometrium, stomach, small intestine, hepatobiliary system, upper urethral tract, larynx and ovary; sebaceous carcinoma, BCC with sebaceous differentiation | Autosomal dominant | |
Other Disorders of Genome Instability | ||||
Bloom syndrome (BS) | Photosensitivity (burning on sun exposure), malar erythema, café-au-lait macules | Most cancer types; in particular leukemia, lymphomas, carcinomas of the breast and gastrointestinal tract | Immune deficiency, growth retardation, unusual facies, male infertility and female subfertility, type II diabetes | Autosomal recessive |
Werner syndrome (WS) | Graying of hair, skin atrophy, leg ulcers, melanomas | Sarcomas, thyroid cancer, meningiomas, melanomas (acral lentiginous, and mucous membrane melanomas) | Premature aging (atherosclerosis, diabetes mellitus, osteoporosis, cataracts) | Autosomal recessive |
Rothmund–Thomson syndrome (RTS) | Photosensitivity (burning on sun exposure), poikiloderma, alopecia | Osteosarcomas, cutaneous SCC and others | Skeletal abnormalities, growth deficiency, juvenile cataracts, osteoporosis | Autosomal recessive |
Fanconi anemia (FA) | Café-au-lait macules | Myeloid leukemia, SCC of head and neck | Aplastic anemia, pancytopenia, growth retardation, thumb and other bone abnormalities | Autosomal recessive |
Dyskeratosis congenita | Lacy, reticular pigmentation of neck and upper chest; nail dystrophy; premature gray hair, hyperhidrosis, skin cancer | Mucosal leukoplakia leading to cancer of anus or mouth, Hodgkin disease, pancreatic adenocarcinoma | Stenosis of lacrimal duct, anemia, pancytopenia, immunodeficiency, learning difficulties, deafness, brain calcifications, cerebellar hypoplasia, testicular atrophy, short stature, intrauterine growth retardation, retinopathy | X-linked recessive, autosomal dominant, autosomal recessive |
Ataxia telangiectasia (AT) | Telangiectasias | T-cell leukemia, lymphomas | Progressive cerebellar ataxia, immune defects, hypogonadism, increased acute toxicity of therapeutic X-ray | Autosomal recessive |
AT-like disorder (ATLD) | Similar to AT | Similar to AT | Similar to AT, but no ocular telangiectasias | Autosomal recessive |
Nijmegen breakage syndrome (NBS) | Café-au-lait macule, vitiligo | B- and T-cell lymphomas, rhabdomyosarcoma, neuroblastoma | Immune defects, growth retardation, microcephaly, mental retardation, characteristic facies | Autosomal recessive |
Seckel syndrome | Café-au-lait macules | Leukemia | Proportionate dwarfism, microcephaly, mental retardation, characteristic facies (receding forehead, narrow face, large beaked nose, micrognathia), immune deficiency, pancytopenia | Autosomal recessive |
Disorder | Mechanisms | ||
|---|---|---|---|
Cellular Abnormalities | Affected Gene(s) | Impaired Function | |
Disorders of Genome Instability with defective DNA repair | |||
Xeroderma pigmentosum (XP) | Increased UV-induced cell killing and mutagenesis | XPA, XPB, XPC, XPD, XPE, XPF, XPG, or DNA polymerase η | Nucleotide excision repair: global genome and transcription-coupled DNA repair; translesional DNA synthesis (XP-variant) |
Cockayne syndrome (CS) | Increased cell killing and mutagenesis after exposure to UV and ionizing radiation | CSA or CSB | Nucleotide excision repair: transcription-coupled DNA repair only |
Xeroderma pigmentosum/Cockayne syndrome complex (XP/CS) | Increased UV-induced cell killing and mutagenesis | XPB, XPD, or XPG | Nucleotide excision repair: global genome and transcription-coupled DNA repair |
Trichothiodystrophy (TTD) | Increased UV-induced cell killing | XPB, XPD, TTD-A, or TTDN1 | Nucleotide excision repair: transcription-coupled pathway of nucleotide excision repair |
Xeroderma pigmentosum/trichothiodystrophy (XP/TTD) | Same as XP | XPD | Nucleotide excision repair: global genome and transcription-coupled DNA repair |
Hereditary nonpolyposis colon cancer (HNPCC)/MUIR–TORRE SYNDROME (MTS) | High level of microsatellite instability in tumors | hMSH2 (MTS, HNPCC1), hMLH1 (MTS, HNPCC2), PMS1 (HNPCC3), hPMS2 (HNPCC4), hMSH6 (HNPCC5), TGFBR2 (HNPCC6), or hMLH3 (HNPCC7) | Mismatch repair |
Other Disorders of Genome Instability | |||
Bloom syndrome (BS) | Spontaneous chromosome breakage and rearrangements, increased sister chromatid exchanges, quadriradial chromosomes | BLM | A RecQ DNA helicase, recombination, replication |
Werner syndrome (WS) | Chromosomal instability, accelerated telomere shortening | WRN | A RecQ DNA helicase, transcription, DNA repair (DSBR, SSBR, recombination), DNA replication, telomere maintenance |
Rothmund–Thomson syndrome (RTS) | Chromosomal instability in response to ionizing radiation | RECQL4 (some patients) | A RecQ DNA helicase |
Fanconi anemia (FA) | Increased sensitivity to DNA cross-linking agents | FANCA, FANCB, FANCC, FANCD1/BRCA2, FANCD2, FANCE, FANCF, FANCG/XRCC9, FANCI, FANCJ/BACH1/BRIP1, FANCL, FANCM, FANCN/PALB2 | DNA damage signaling, recombination repair |
Dyskeratosis congenita | Shortened telomeres | DKC1 X-linked; TERC, TERT, TINF2, autosomal dominant; TERT, NOLA3, NOLA2 autosomal recessive | Abnormal telomere biology |
Ataxia telangiectasia (AT) | Increased spontaneous and X-ray-induced chromosome breakage, increased sensitivity to killing by ionizing radiation, impaired cell cycle arrest and/or apoptosis in response to DNA damage | ATM | DNA damage signaling (control of cell cycle, apoptosis), repair of DNA double strand breaks |
AT-like disorder (ATLD) | Increased spontaneous and X-ray-induced chromosome breakage, increased sensitivity to killing by ionizing radiation, impaired DNA strand break repair | LIG4 | DNA ligase 4, strand break repair |
Nijmegen breakage syndrome (NBS) | Increased spontaneous and X-ray-induced chromosome breakage, increased sensitivity to killing by ionizing radiation | NBS1 | Nibrin (p95), which is part of the MRE11–RAD50–NBS1 complex, DNA damage signaling, recombination, DNA double strand break repair, cell cycle checkpoint |
Seckel syndrome | Spontaneous and mitomycin C- induced chromosome instability, sensitivity to killing by UV and DNA cross-linking agents | ATR | Ataxia telangiectatica- and RAD3-related protein, DNA damage response, cell cycle checkpoints |
In addition, although these heritable diseases are rare (of the order of 10−5 or 10−6), carriers of the affected genes may comprise several percent of the general population. These individuals are usually free of clinical symptoms, as most of these disorders are characterized by autosomal recessive inheritance. However, epidemiologic studies suggest that heterozygotic carriers may have an increased risk of neoplasia as well.5,6
Spontaneous genome instability is present in Bloom syndrome (BS), ataxia telangiectasia, and Fanconi anemia (FA) as manifested by increased chromosome breakage in primary blood or skin cells. On the other hand, genome instability is present in cells from patients with xeroderma pigmentosum (XP) only after exposure to DNA-damaging agents such as UV radiation or other carcinogens such as benzo[a]pyrene, which is present in cigarette smoke. In XP, many of the severe disease manifestations such as cancer and corneal scarring leading to blindness are the result of the interplay between genetic risk and environmental exposure. For example, XP patients who avoid UV radiation dramatically reduce or eliminate the probability of developing skin cancer and blindness.
In some of the disorders discussed here, genome instability is caused by an impaired ability to repair damage to DNA introduced by certain physical or chemical agents. Cells are equipped with different DNA repair pathways (reviewed in Chapter 110) that repair different types of DNA damage. The nucleotide excision repair (NER) pathway, which processes bulky DNA lesions, including UV-induced DNA photoproducts, is impaired in XP, Cockayne syndrome (CS), and trichothiodystrophy (TTD). Cells from patients with these conditions are characterized by increased killing and mutation formation following exposure to UV radiation. However, only XP patients have increased cancer risk. The reason for this difference in cancer risk is not known.
Proteins encoded by some of the affected genes in these three disorders are not only involved in DNA repair, but also in transcription and cellular DNA damage responses. Thus, mutations affecting different functions of the same gene (such as nervous system development or immune competence) appear to play a role in various phenotypes that are not directly linked to DNA repair functions and underlie several overlap syndromes between those three conditions (Table 139-3). Multiple clinical phenotypes of DNA repair diseases have been recognized. Defects in many genes have been associated with these clinical phenotypes. Mutations in different DNA repair genes can be associated with the same clinical phenotype. On the other hand, different mutations in one DNA repair gene can be associated with different clinical phenotypes. For example, mutations in the XPD(ERCC2) gene have been associated with the following clinical phenotypes: XP, XP with neurologic abnormalities, the XP-Cockayne syndrome complex (XP/CS), XP/TTD, COFS (cerebro-oculofacio-skeletal syndrome), or CS/TTD. XP variant, which is clinically indistinguishable from XP, is a disorder of DNA damage tolerance. Although normal cells can bypass DNA damage during replication (translesional DNA synthesis), cells from XP-variant patients have a defect in this process. The DNA mismatch repair pathway (see Chapter 110) is affected in hereditary nonpolyposis colon cancer (Lynch syndrome) and its subform, Muir–Torre syndrome. Muir-Torre syndrome has cutaneous features including sebaceous gland tumors. These are reviewed in Chapter 153.
Clinical Disorders | Molecular Defects |
|---|---|
Xeroderma pigmentosum | XP-C, XP variant, XP-D, XP-A, XP-F, XP-G, XP-E |
Xeroderma pigmentosum with neurological Abnormalities | XP-A, XP-D, XP-B |
Trichothiodystrophy | XP-D, TTD-A, XP-B, TTDN1 |
Cockayne syndrome | CS-B, CS-A |
Xeroderma pigmentosum/Cockayne syndrome complex | XP-B, XP-G, XP-D |
Xeroderma pigmentosum/Trichothiodystrophy complex | XP-D |
| Cerebro-ocular-facial skeletal (COFS) syndrome | CS-B, XP-D |
Helicases are proteins that unwind DNA and are required for a multitude of metabolic processes involving DNA, such as transcription, replication, repair, and more. Two genes involved in NER are helicases that are also part of the basal transcription factor, TFIIH. The XPB (ERCC3) gene product unwinds DNA in the 3′ to 5′ direction whereas the XPD (ERCC2) gene product unwinds DNA in the opposite direction. The RecQ family of DNA helicases is highly conserved in evolution from bacteria to humans.7,8 Of the five known human RecQ family members, three [(1) BLM, (2) WRN, and (3) RECQL4, which cause BS, Werner syndrome (WS), and Rothmund–Thomson syndrome (RTS), respectively] are mutated in distinct clinical disorders associated with chromosomal instability, cancer predisposition, and/or premature aging.9
Genetic instability can also result from abnormal telomere maintenance. Telomeres are long nucleotide (TTAGGG)n repeats that are important for the maintenance of chromosomal integrity.10 Some telomeric repeats are lost at each cell division and shortened telomere lengths are a feature of aged cells. Accelerated telomere shortening is a hallmark of cells from patients with the cancer-prone disorder dyskeratosis congenita. In about half of dyskeratosis congenita patients, mutations have been found in one out of six genes involved in telomere maintenance.11
BASC (BRCA1-associated genome surveillance complex) is a multienzyme complex centered around the BRCA1 protein in the nucleus.12 It contains important DNA damage response proteins including ATM, ATR, the MRE11-RAD50-NBS1 complex, BRCA1, BLM, FANCD2, and MLH1. These proteins physically interact with each other. Genome instability may result from a defect in any of those and cause the genome instability disorders ataxia-telangiectasia (reviewed in Chapter 143), Seckel syndrome, Nijmegen breakage syndrome, hereditary breast cancer, BS, FA, or hereditary colon cancer. BS and FA are reviewed here.
Disorders of DNA Repair
XP serves as the prototype heritable disease with increased sensitivity to cellular injury.12–14 XP is an autosomal recessive disease with sun sensitivity, photophobia, early onset of lentigines and freckling, and subsequent neoplastic changes on sun-exposed surfaces. There is cellular hypersensitivity to UV radiation and to certain chemicals in association with abnormal DNA repair. Some of the patients have progressive neurologic degeneration.
XP occurs with a frequency of about 1 in 1 million persons in Europe and the United States15,16 It is relatively more common in areas such as the Middle East where marriage of close relatives is practiced. Patients have been reported worldwide in all races, including whites, Asians, blacks, and Native Americans.
Approximately one-half the patients with XP have a history of acute sunburn reaction on minimal UV exposure. The other patients tan normally without excessive burning. In all patients, numerous freckle-like hyperpigmented macules (lentigines) appear predominately on sun-exposed skin (Fig. 139-1). The median age of onset of the cutaneous symptoms in XP is between 1 and 2 years (Fig. 139-2).14 These generally spare sun-protected sites such as the buttocks (see Fig. 139-1D). However, some severely sun-exposed patients may show pigmentary abnormalities in the axillae. Continued sun exposure causes the patient’s skin to become dry and parchment-like, with increased pigmentation, hence the name xeroderma pigmentosum (“dry pigmented skin”; see Fig. 139-1A). Premalignant actinic keratoses develop at an early age (see Fig. 139-1B). The appearance of sun-exposed skin in children with XP is similar to that occurring in farmers and sailors after many years of extreme sun exposure.
Figure 139-1
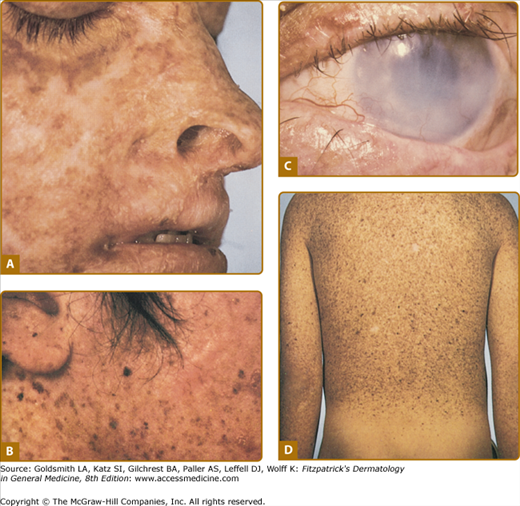
Xeroderma pigmentosum. A. Pigmentary changes, atrophy, dryness, and cheilitis in a 16-year-old patient. B. Cheek of a 14-year-old patient with pigmented macules of varying size and intensity, actinic keratosis, basal cell carcinoma, and a surgical scar. C. Corneal clouding, prominent conjunctival blood vasculature, and loss of lashes. D. Myriads of pigmented macules of varying size and intensity and scattered achromic areas on the back, with marked sparing of sun-protected buttocks of a 14-year-old patient.
Figure 139-2
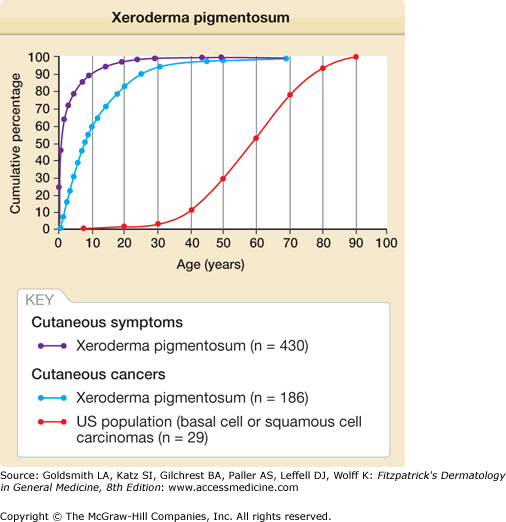
Xeroderma pigmentosum. Age at onset of clinical symptoms and skin cancer. Age at onset of cutaneous symptoms (generally sun sensitivity or pigmentation) was reported for 430 patients. Age at first skin cancer was reported for 186 patients and is compared with the age distribution for 29,757 patients with basal cell carcinoma or squamous cell carcinoma in the US general population. There is a 50-year reduction in age of onset of skin cancer in the XP patients compared with the US general population. (From Kraemer KH, Lee MM, Scotto J: Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol 123:241, 1987, with permission.)
Patients with XP younger than 20 years of age have a greater than 10,000-fold increased risk of cutaneous basal cell carcinoma, squamous cell carcinoma, or melanoma.12,13,16,17 The median age of onset of nonmelanoma skin cancer reported in patients with XP is 8 years. This 50-year reduction in comparison with the general population is an indication of the importance of DNA repair in protection from skin cancer in unaffected individuals (see Fig. 139-2).
Review of the world’s literature on XP has revealed a substantial number of cases of oral cavity neoplasms, particularly squamous cell carcinoma of the tip of the tongue, a presumed sun-exposed location. Brain (sarcoma and medulloblastoma), central nervous system (astrocytoma of the spinal cord), lung, uterine, breast, pancreatic, gastric, renal, and testicular tumors and leukemia have been reported in a few patients with XP.11,12,15,16 Overall, these reports suggest an approximate 10- to 20-fold increase in internal neoplasms in XP.
Ocular abnormalities are almost as common as the cutaneous abnormalities and are an important feature of XP (see Fig. 139-1C).14,18,19 The posterior portion of the eye (retina) is shielded from UV radiation by the anterior portion (lids, cornea, and conjunctiva). Clinical findings are strikingly limited to these anterior, UV-exposed structures. Photophobia is often present and may be associated with prominent conjunctival injection. Schirmer’s testing frequently reveals reduced tearing leading to dry eyes. Continued UV exposure of the eye may result in severe keratitis, leading to corneal opacification and vascularization. The lids develop increased pigmentation and loss of lashes. Atrophy of the skin of the lids results in ectropion, entropion, or, in severe cases, complete loss of the lids. Benign conjunctival inflammatory masses or papillomas of the lids may be present. Epithelioma, squamous cell carcinoma, and melanoma of UV-exposed portions of the eye are common. The ocular manifestations may be more severe in black patients.20
Neurologic abnormalities have been reported in approximately 30% of the patients.13,21 The onset may be early in infancy or, in some patients, delayed until the second decade. The neurologic abnormalities may be mild (e.g., isolated hyporeflexia) or severe, with progressive mental retardation, sensorineural deafness (beginning with high-frequency hearing loss), spasticity, or seizures. The most severe form, known as the De Sanctis–Cacchione syndrome, involves the cutaneous and ocular manifestations of classic XP plus additional neurologic and somatic abnormalities, including microcephaly, progressive mental deterioration, low intelligence, hyporeflexia or areflexia, choreoathetosis, ataxia, spasticity, Achilles tendon shortening leading to eventual quadriparesis, dwarfism, and immature sexual development. The complete De Sanctis–Cacchione syndrome has been recognized in very few patients; however, many patients with XP have one or more of its neurologic features. In clinical practice, deep tendon reflex testing and routine audiometry usually can serve as a screen for the presence of XP-associated neurologic abnormalities. In cases where there is clinical evidence of early neurologic abnormalities, a brain magnetic resonance imaging (MRI) may show enlarged ventricles.
The predominant neuropathologic abnormality found at autopsy in patients with neurologic symptoms was loss (or absence) of neurons, particularly in the cerebrum and cerebellum. There is evidence for a primary axonal degeneration in these patients.20 In a long term follow-up study of 106 XP patients those with neurodegeneration had a younger age at death (29 years) than those without neurodegeneration (37 years).
Cultured cells from patients with XP generally grow normally when not exposed to damaging agents. However, the population growth rate or single-cell colony-forming ability is reduced to a greater extent than normal cells after exposure to UV radiation. A range of post-UV colony-forming abilities has been found with fibroblasts from patients, some having extremely low post-UV colony-forming ability and others having nearly normal survival (see Chapter 110).12,14
XP fibroblasts are deficient in their ability to repair some UV-damaged viruses or plasmids to a functionally active state.12,14,20 These host cell reactivation assays have detected an abnormality in every form of XP tested.
UV-irradiated XP fibroblasts are hypermutable compared to normal fibroblasts. This post-UV hypermutability is believed to be the basis of the increased frequency of sunlight-induced somatic mutations that lead to cancer in XP patients.22
XP cells generally are found to have a normal karyotype without excessive chromosome breakage or increased sister chromatid exchanges (as seen in BS). However, after exposure to UV radiation, abnormally large increases in chromosome breakage and in sister chromatid exchanges have been observed.23 The extent of this induced abnormality varies in different patients.
In 1968, hypersensitivity of cultured XP cells to UV damage was reported by Cleaver24 to be the result of defective DNA repair. He found defective UV-induced repair replication, indicating a defect in the NER pathway. Most XP cells have a normal response to treatment with X-rays, indicating the specificity of the DNA repair defect.25 The defective genes for the seven NER-defective forms of XP and the XP variant have been cloned26 and their functions are being investigated (see Chapter 110).
Genetic heterogeneity among the XP DNA repair defects was found by fusing cultured fibroblasts from different patients and defining complementation groups (see Chapter 110). Up to 2011, seven such DNA excision repair-deficient complementation groups have been identified (named XP-A to XP-G) and the corresponding genes have been identified (see Table 139-3).26 Additional patients with clinical XP but normal NER have been called XP variants. Studies of cellular hypersensitivity revealed a slightly increased sensitivity to UV-induced inhibition of cell growth that was potentiated by caffeine. Cells from XP-variant patients have a defect in an error-prone DNA polymerase (pol η) that bypasses unrepaired DNA damage.27,28
Prenatal diagnosis has been reported by measuring UV-induced unscheduled DNA synthesis in cultured amniotic fluid cells29 and by use of DNA diagnosis of trophoblast cells obtained early in pregnancy.30,31 DNA-based prenatal diagnosis may also be possible in selected cases.32
A number of DNA-damaging agents other than UV radiation have been found to yield hypersensitive responses with XP cells. These agents include drugs (psoralens, chlorpromazine), cancer chemotherapeutic agents (cisplatin,33 carmustine), and chemical carcinogens (benzo[a]pyrene derivatives). Presumably, these agents induce DNA damage whose repair involves portions of the DNA repair pathways that are defective in XP.33
Management of patients with XP is based on early diagnosis, lifelong protection from UV radiation exposure, and early detection and treatment of neoplasms. Diagnosis rests on recognition of the characteristic clinical features and is confirmed by laboratory tests of cellular hypersensitivity to UV and defective DNA repair (see Chapter 110). Molecular determination of some of the XP disease-causing mutations is offered in a laboratory that is certified for clinical testing (see http://genetests.org for the most recent listing).
Patients should be educated to protect all body surfaces from UV radiation by wearing protective clothing and UV-absorbing glasses and long hair styles. They should adopt a lifestyle to minimize UV exposure and use sunscreens with high sun protective factor (SPF) ratings (minimum SPF 30) daily. Patients should be examined frequently by a family member who has been instructed in recognition of cutaneous neoplasms. A set of color photographs of the entire skin surface with close-ups of lesions (including a ruler) is often extremely useful to both the patient and the physician in detecting new lesions. A physician should examine patients at frequent intervals (approximately every 3–6 months depending on severity of skin disease). Premalignant lesions such as actinic keratoses may be treated by freezing with liquid nitrogen, or with topical 5-fluorouracil or imiquimod. Photodynamic therapy, using, for example, the topical photosensitizer 5-aminolevulinic acid followed by exposure to blue light, is an effective treatment modality for normal patients with multiple actinic keratoses. There are no data on the safety or efficacy of this treatment in XP patients. Caution is recommended because an abnormal response to photodynamic therapy or other light- or laser-based therapies cannot be excluded in XP cells. Larger areas have been treated with therapeutic dermatome shaving or dermabrasion to remove the more damaged superficial epidermal layers.34–37 This procedure permits repopulation by relatively UV-shielded cells from the follicles and glands.38
Because cells from patients with XP are also hypersensitive to environmental mutagens such as benzo[a]pyrene found in cigarette smoke, prudence dictates that patients should be protected against these agents. One of our patients who smoked cigarettes for more than 10 years died of bronchogenic carcinoma of the lungs at age 35 years and another patient who smoked has developed a lung cancer at age 48 years.39 Thus, we recommend that XP patients refrain from smoking cigarettes and that parents should protect children with XP from being exposed to secondhand smoke.
Cutaneous neoplasms are treated in the same manner as in patients who do not have XP. This involves electrodesiccation and curettage, surgical excision, or Mohs micrographic surgery (see Chapters 115 and 244). Because multiple surgical procedures are often necessary, removal of undamaged skin should be minimized. Extremely severe cases have been treated by excision of large portions of the facial surface and grafting with uninvolved skin.35
Most patients with XP are not abnormally sensitive to therapeutic X-rays, and XP patients have responded normally to full doses of therapeutic X-radiation for treatment of inoperable neoplasms such as an astrocytoma of the spinal cord,40 a frontal lobe astrocytoma, or recurrent squamous cell carcinoma in the orbit. However, cultured cells from two XP patients were found to be hypersensitive to X-rays,41,42 so when X-ray therapy is indicated, an initial small dose is advisable to test for clinical hypersensitivity.
Oral isotretinoin has been shown in a controlled study to be effective in preventing new neoplasms in patients with multiple skin cancers.43 Because of its toxicity (hepatic, hyperlipidemic, teratogenic, calcification of ligaments and tendons, premature closure of the epiphyses), oral isotretinoin should be reserved for patients with XP who are actively developing large numbers of new skin cancers. We found that the effective dose varies among patients and some patients may respond to doses of oral isotretinoin as low as 0.5 mg/kg/day.
A bacterial DNA repair enzyme, denV T4 endonuclease, in a topical liposome-containing preparation, has been reported to reduce the frequency of new actinic keratoses and basal cell carcinomas in XP patients in one research study.44 As of 2010, this treatment has not been approved by the U.S. Food and Drug Administration.
A study treating multiple melanoma in-situ lesions with intralesional interferon-α in one XP patient showed localized clearing only of lesions injected with the intralesional interferon-α but not with the control diluent.45 There are several case reports of XP patients responding to topical treatment with the immune modulator imiquimod (see Chapter 221).46–49 However, none of these reported long-term follow-up.
The eyes should be protected by wearing UV-absorbing glasses with side shields. Methylcellulose eye drops can be used to keep the cornea moist. Corneal transplantation has restored vision in patients with corneal opacity from severe keratitis. However, some of these suffered graft rejection due to neovascularization. Neoplasms of the lids, conjunctiva, and cornea are usually treated surgically.50–52 We are examining the possibility of using a swab to obtain cytologic specimens from the surface of the eye to determine if early neoplasms can be detected or excluded without the need for performing biopsies.
Patients with XP are hypersensitive to UV radiation, as are their cultured cells. Cutaneous and ocular abnormalities are strikingly limited to UV-exposed areas and usually spare such UV-shielded locations as the axillae, buttocks, and retina. The fact that black patients with XP have an increased frequency of skin cancer suggests that a normally functioning DNA repair system provides greater protection against skin cancer than does the natural pigmentation of black skin.
At least eight different molecular defects are associated with the clinical abnormalities recognized as XP, as indicated by the existence of seven DNA excision repair-deficient complementation groups (A to G) and the variant form. A discussion of the cloned XP genes and their function can be found in Chapter 110. A Web site listing disease-causing mutations in XP and CS genes has been established at http://xpmutations.org/. There is a complex relationship among the DNA repair genes and clinical disease (see Table 139-3 and see http://genetests.org for review of xeroderma pigmentosum). Multiple NER genes are associated with at least a spectrum of different clinical phenotypes. A clinical phenotype can be associated with defects in each of several genes. Conversely, mutations in one gene can be associated with several different clinical phenotypes. These complex relationships and the roles of DNA repair genes in regulation of transcription and in immune functions are under intense investigation.
Complementation group A (see Table 139-3) contains patients with the most severe neurologic and somatic abnormalities (the De Sanctis–Cacchione syndrome) as well as patients with minimal or no neurologic abnormalities.13 Long-term follow-up of these patients has revealed a relationship between the genotype and the phenotype. Patients with the most severe disease appear to have truncating mutations in both alleles of the XPA gene leading to no detectible normal protein. In contrast, patients with minimal neurologic abnormalities have splice-site mutations that permit a small amount of normal messenger RNA (mRNA) to be made. This form is seen in the United States, Europe, and the Middle East. It is the most common form of XP in Japan. Approximately 90% of Japanese XP-A patients have the same single-base-substitution founder mutation.53 This finding has served as the basis for development of a rapid diagnostic assay for Japanese XP-A patients (including prenatal diagnosis) using polymerase chain reaction analysis of a small sample of DNA.37 Heterozygous carriers of this disease-causing mutation who have one mutated allele and one normal allele have been estimated to comprise approximately 1% of the Japanese population.16
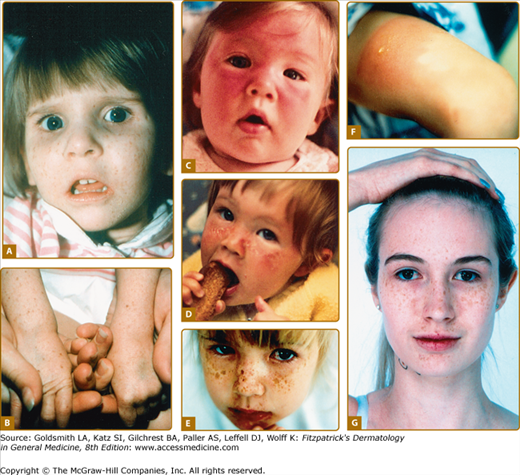
Complementation group B (Fig. 139-3) is composed of five patients in four kindreds who had the cutaneous abnormalities characteristic of XP (including neoplasms) in conjunction with neurologic and ocular abnormalities typical of CS.13,54 Another family had two adult sisters with XP without CS who had ocular melanomas and were parents of normal children. Surprisingly, a patient with TTD also was found to have a defect in the XPB gene.
Figure 139-3

Xeroderma pigmentosum (XP/CS) complex. A. 28-year-old patient (XP11BE) in XP complementation group B with cutaneous changes of XP, including pigmentary changes and atrophy. She has a beak-like nose and loss of subcutaneous tissue typical of Cockayne syndrome. B. The patient is of short stature (less than 4 ft. tall). Her mother, an obligate heterozygote, is clinically normal.
Patients in complementation group C, with rare exceptions, have XP with skin and ocular involvement but without neurologic abnormalities.13,55–61 This is the most common group in the United States, Europe, and Egypt, but has been found rarely in Japan. Most patients have truncating mutations in both alleles leading to undetectable levels of XPC mRNA (due to nonsense-mediated message decay). However, a splice lariat branchpoint mutation resulting in as little as 3% to 5% of normal mRNA resulted in milder clinical symptoms in one family in Turkey.58 XP-C patients typically do not give a history of severe blistering sunburns on minimal sun exposure and at times are first diagnosed with the appearance of skin cancer in a child. One XP-C patient was reported to be hypersensitive to ionizing radiation42; however, correction of the XPC gene defect did not correct the cellular ionizing radiation hypersensitivity, suggesting that more than one gene was defective in this patient.
Patients in complementation group D have been described with several different clinical phenotypes. They may have cutaneous XP with late onset of neurologic abnormalities in their second decade of life or XP with no neurologic abnormalities.13,62 Two XP-D patients have been reported with clinical symptoms of both XP and CS. Cells from patients with a photosensitive form of TTD (without XP) also were assigned to the XP complementation group D. Two patients were reported with combined symptoms of both TTD and XP (one patient had a skin cancer and another died of metastatic melanoma) and mutations in the XPD gene.63 Finally, a patient with cerebro-oculo-facio-skeletal (COFS) syndrome had a mutation in the XPD gene.64
Complementation group E was found in one kindred in Europe and several in Japan.13,65 We have studied adult patients with multiple skin cancer in 3 kindreds in the US and Germany. These patients had no neurologic involvement.66
Complementation group F patients have been found mainly in Japan.67,68 Most of these patients have mild clinical symptoms without neurologic abnormalities or skin cancer. However, we recently found two families with adult onset of severe neurodegeneration with mutations in the XPF gene. The residual rate of DNA repair is very low (only 10% to 20% of normal).
Thirteen patients in XP complementation group G have been identified in the United States, Europe, and Japan (Fig. 139-4).69 There is a large variation in clinical features among these patients. Several patients with mutations in the XPG gene had clinical features of both XP and severe CS with cachexia and death in the first decade (see Fig. 139-3). Other patients with different mutations in the same gene had no neurologic abnormalities.
Figure 139-4
Severe and mild xeroderma pigmentosum XP-G patients. A and B. XP82DC. C–F. XP65BE. A. Patient XP82DC at 3 years of age has deep-set eyes characteristic of Cockayne syndrome (CS) and irregular lentiginous pigmentation on her face characteristic of XP, indicating XP/CS complex. B. Patient XP82DC at 3 years of age has characteristic XP-pigmented lesions on her forearms and dorsa of hands along with thin, translucent skin with readily visible veins. The small size of her hands is apparent in comparison with the hands of her mother. C. Patient XP65BE at age 6 months experienced severe sunburn on her face with minimal sun exposure. Erythema and swelling are seen on skin of forehead, cheeks, and periorbital area. D. Patient XP65BE at age 9 months shows erythema and peeling of skin of malar area of face after sun exposure. E. Patient XP65BE at age 4.5 years shows pigmentary changes on her nose, malar area, and other portions of her face. F. Patient XP65BE at 4.5 years shows blistering sunburn on her upper thigh. Note spared area above knee where sunscreen was applied G. Patient XP65BE shows minimal pigmentary changes on face and sparing of neck and hand. She used measures to protect her skin from sun exposure. (From Emmert S et al: Relationship of neurologic degeneration to genotype in three xeroderma pigmentosum group G patients. J Invest Dermatol 118:972, 2002, with permission.)
XP-variant cells have normal DNA NER and thus do not fall into any of the complementation groups of cells with defective DNA excision repair.14 However, there is a defect in an error-prone, translesional DNA damage bypass polymerase, pol η (see Chapter 110).25,27,70 Most XP-variant patients have clinical XP with no neurologic abnormalities.71 The cutaneous and ocular abnormalities have been severe in some patients and mild in others.
XP heterozygotes (parents and some other relatives) are carriers of the gene for XP but are clinically normal. There is limited epidemiologic evidence to indicate that such persons have an increased risk of developing skin cancer.72 Most tests of cell function or DNA repair yield normal responses with cells from XP heterozygotes.
The Xeroderma Pigmentosum Society is an educational, advocacy, and support organization for XP patients and their families: Xeroderma Pigmentosum Society, Inc., Box 4759, Poughkeepsie, NY 12602–4759; Web site: http://www.xps.org; e-mail: xps@xps.org; telephone: toll-free (877) XPS-CURE (877-977-2873). Another support group is the XP Family Support Group, 8375 Folsom Blvd Suite 201, Sacramento Ca, 95826. Their Web site is http://www.xpfamilysupport.org/. A Web site listing disease-causing mutations in XP and CS genes has been established at http://xpmutations.org/.
CS is a very rare, autosomal recessive degenerative disease with cutaneous, ocular, neurologic, and somatic abnormalities (see Table 139-1).73,74 A review published in 1992 described 140 cases reported in the literature.75
In 1936, E. A. Cockayne described a syndrome characterized by cachectic dwarfism, deafness, and pigmentary retinal degeneration with a characteristic “salt and pepper” appearance of the retina.76 The skin had photosensitivity without the excessive pigmentary abnormalities seen in XP. There was marked loss of subcutaneous fat, resulting in a “wizened” appearance with typical “bird-headed” facies and prominent “Mickey Mouse” ears. Other ocular findings included cataracts and optic atrophy.77 Neurologic abnormalities, in addition to deafness, include peripheral neuropathy, normal pressure hydrocephalus, and microcephaly. Birth weight and early development are usually normal. The disease onset is usually in the second year of life with slowly progressive neurologic degeneration. Intellectual deterioration may be nonuniform, with some functions preserved better than others. A severe infantile form has been described78,79 as well as a milder form with late onset. COFS syndrome64,80,81 with microcephaly and severe mental retardation and CAMFAK syndrome of congenital cataracts, microcephaly, failure to thrive, and kyphoscoliosis82 have some similar features. CS is not associated with an increased incidence of neoplasia.
Clinical laboratory testing often shows sensorineural deafness, neuropathic electromyogram, and slow motor nerve conduction velocity.13,20,69,75,78,83 The electroencephalogram may be abnormal, and X-ray examination may show thickened skull and microcephaly. Computed tomography may be diagnostically useful in the detection of normal-pressure hydrocephalus and showing calcification of the basal ganglia and other structures (see eFig. 139-4.1
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


