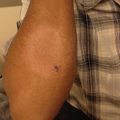
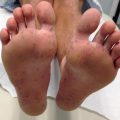
Fig. 13.1
Clinical presentation of patients with Epidermolysis Bullosa. (a) Typical blistering leading to chronic wounds in a patient with RDEB. (b) Blisters on the foot at the sites of trauma in a patient with EBS
Blistering occurs more commonly at sites of friction (flexural and extensor surfaces)
In EBS Dowling-Meara, blisters occur in characteristic clusters (herpetiform), and involvement of mucous membranes including the mouth, larynx, and esophagus is common
Healing is typically without scarring
Intra-oral involvement may occur in generalized type of EBS
Nail dystrophy is rare
Improvement of the blistering may occur with age
Junctional EB (JEB)
is inherited in an autosomal recessive manner. This is a heterogeneous disorder that results from mutations in proteins present at the DEJ (plectin, laminin 332, collagen XVII and α6β4 integrin). These are all components of the anchoring filaments and hemidesmosomes. Subtypes include JEB-Herlitz, JEB non-Herlitz, JEB with pyloric atresia, JEB inversa and laryngo-onycho-cutaneous syndrome (also known as Shabbir syndrome).
Clinical Features of JEB
Tense bullae on the skin and extensive oral mucous membranes involvement at birth
Involvement of the buttocks and pinnae, periorificial and subungal involvement is common in Herlitz-JEB
Nail dystrophy, dental enamel hypoplasia, non-scarring alopecia, and conjunctival involvement are common
JEB-Herlitz is associated with increased risk of sepsis, airway compromise, and death in the first few years after birth. Patients with JEB non-Herlitz have a milder phenotype, but it may sometimes be indistinguishable from JEB-Herlitz, especially in infancy. Patients with JEB non-Herlitz survive into adulthood.
Dystrophic EB (DEB)
can be dominantly (DDEB) or recessively (RDEB) inherited. In general, DDEB has a milder phenotype than RDEB. Mutations of genes encoding type VII collagen (COL7A1), present in anchoring fibrils, result in DEB [1].
Clinical Features of DEB (See Fig. 13.1)
Tense blisters/bulla at the sites of trauma, healing with scarring
Various degrees of mucous membranes involvement, with potential for scarring, depending on the subtype (e.g. esophageal strictures, corneal synechiae, etc.)
Extensive nail and digit involvement leading, in severe cases, to mitten deformity
Increased likelihood of squamous cell carcinoma
Severe cases have complications (e.g. anemia, malnutrition, cardiomyopathy, renal involvement, osteoporosis, etc.)
Specific Strategies
Management strategies involve prompt recognition and diagnosis of EB and its subtype, and supportive care including, but not limited to, blister prevention, wound care, and prevention of complications.
Investigations Recommended
For diagnosis |
Skin biopsy for immunofluorescence antigenic mapping |
Skin biopsy for transmission electron microscopy (ultrastructural analysis) |
Skin biopsy for light microscopy |
Genetic testing |
Intrauterine skin biopsy (prenatal diagnosis) |
Light microscopy is not useful for establishing the subtype of EB, but can help exclude other vesiculobullous diseases.
A biopsy should be taken from an unblistered area of skin, which has been gently rubbed with a pencil eraser for 1 min (this is to create a cleavage in the skin at a microscopic level). A shave biopsy is preferred, as it provides a longer specimen of the DEJ. The specimen is then gently cut in half for ultrastructural and immunohistochemical analysis. Punch biopsies should be avoided, as the rotational action may cause complete separation of the epidermis from the dermis.
Immunofluorescence mapping (IFM), when coupled with the use of specific monoclonal antibodies, can provide considerable insight into the major type of EB and the structural protein most likely mutated. In many centres, this testing can provide fast and reliable subtype diagnosis and can inform on the genetic panel that needs to be requested [2].
Electron microscopy (EM) is the gold standard for determining the cleavage plane, however, it may be cost prohibitive. The main advantage of EM is that it also allows visualization and semi-quantitative assessment of specific structures of the BMZ. Due to very few recommended reference laboratories worldwide, EM is likely to have a decreasing role in the diagnosis of EB in the future, although it is likely to continue to have an important place in research [3]. In EBS the cleavage plane is either at the infra-nuclear portion of the basal keratinocytes or in the suprabasal epidermis. In JEB the cleavage plane occurs within the lamina lucida (hemidesmosomes), and in DEB the cleavage occurs immediately beneath the lamina densa (anchoring fibrils).
Genetic analysis often does not affect management of the patient, but aids in determining inheritance pattern of the condition, and allows prenatal testing. This can be done by a prenatal/uterine skin biopsy in the second trimester. Newer methods include chorionic villus sampling or amniocentesis in affected families, and can be undertaken in the first trimester of pregnancy.
Specific Therapies
By and large, the management therapy in EB is supportive, consisting of:
Prevention of new blisters/avoidance of trauma. It is very important to puncture and drain large bullae in order to prevent extension and supra-infection of lesions. Parents of young babies need to be taught how to handle the baby [4], to use clothing inside out or with no seams, and to place babies on mattresses that diminish chances of friction. Foam pads can be sewn into the lining of clothing, especially over bony prominences, and babies/infants should be gently handled to prevent blistering. In older patients and adults, particularly those with EBS types, keeping soles and feet cool and dry, use of topical antiperspirants (20 % aluminium chloride hexahydrate) for hyperhidrosis of acral areas, and use of well-fitting footwear may be beneficial in preventing blistering. More severe subtypes will benefit from foam dressings that offer some padding/protection of unaffected to freshly wounded or healed skin.
Prevention and treatment of infections and promotion of wound healing. There is no evidence that routine use of topical antibiotics reduces the risk of infection or enhances healing. However, painful or exudative wounds may benefit from local antimicrobials (either as topical antibiotics or dressings with antimicrobials, such as silver or polyhexamethylene biguanide, PHMB). Occasionally, systemic antibiotics are needed. In patients with EBS there is some evidence that oral tetracycline decreases the blistering, particularly in the summer months [5]. Similarly, patients with RDEB may experience enhanced healing while on trimethoprim [6]. Wound healing is further enhanced if the patient has adequate nutrition and a hemoglobin level at least of 100 g/L [7].
Education and psychosocial support. Providing information about the condition and its potential complications is extremely important. Patients and families also need psychological support through specialized practitioners and from non-profit organizations such as the National EB registry (NEBR) or DEBRA (dystrophic Epidermolysis Bullosa Research Association).
Multidisciplinary team involvement. EB is, in severe cases, a multisystem disease. As such, many knowledgeable specialists are required to manage the cutaneous and extracutaneous complications of this rare condition (dermatologists, nurses, plastic surgeons, pediatricians, gastroenterologists, hematologists, ophthalmic surgeons, dentists, dieticians, physiotherapists, etc.).
Table 13.2
Second line therapies
Modality | Dosage | Level of evidence |
|---|---|---|
Tetracycline | 1.5 g per day divided BID for 4 months | D |
Trimethoprim | 4 mg/kg/day divided into BID for 2 months | D |
Bone marrow transplantation | D |
A two-arm study with 12 patients given oral tetracycline versus placebo found that there was a definite reduction of blisters in those treated with tetracycline. This, however, was not statistically significant. It is recommended that empirical treatment with oral tetracycline can be given in any EBS patient who is sufficiently symptomatic and willing to risk the adverse effects of oral tetracycline [6].
In a proof-of-concept study involving ten patients with RDEB, six out of seven patients had a 50 % reduction in chronic wound surface area while on trimethoprim versus two out of six patients on placebo. While this was not statistically significant, it provided useful information for further prospective studies [7].
Six patients with RDEB were treated with immunomyeloablative chemotherapy and allogenic stem-cell transplantation. All had improved wound healing and a reduction in blister formation at 30–130 days post-transplantation. Increased collagen VII deposition in five patients and a sustained presence of donor cells were found in all six by immunofluorescence [8].
Epidermolysis Bullosa Acquisita (EBA)
EBA is a chronic subepidermal immunobullous disorder that is rare in children. Three phenotypes have been recognized [9]:
- 1.
Non-inflammatory type that resembles the inherited form of dystrophic EB, with tense bullae on extensor surfaces and at sites of trauma, milia, atrophic scars, pigmentary changes, and nail dystrophy.
- 2.
Inflammatory type with pruritic tense bullae on normal, erythematous or urticarial skin, including sites that are not exposed to trauma.
- 3.
Mucous membrane pemphigoid-like with involvement of conjunctival, oral, nasopharyngeal, and genital mucous membrane, leading to scarring.
The target antigen is type VII collagen. The long-term prognosis in children is good, with remission usually achieved within 1–4 years [10].
Management Strategies
Management strategies include prompt diagnosis and initiation of first line therapies to control the blistering.
Investigations
For diagnosis |
Skin biopsy (lesional) for routine light microscopy |
Skin biopsy (perilesional) for direct immunofluorescence (DIF) |
Serum for indirect immunofluorescence (IIF) |
Immunoelectron microscopy |
Histology of a fresh blister shows a subepidermal bulla and a predominantly neutrophilic infiltrate with eosinophils.
DIF of perilesional skin shows linear deposits of IgG and C3 along the BMZ and weak staining for IgA and IgM.
IIF usually shows the circulating antibody on the dermal side of the bulla of salt-split skin.
Direct immunoelectron microscopy shows IgG deposits under the lamina densa within the region normally occupied by anchoring fibrils [10].
Table 13.3
First line therapies
Modality | Dosage | Level of evidence |
|---|---|---|
Systemic corticosteroids (prednisolone) | 1 mg/kg/day | D |
Dapsone | 1–2 mg/kg/day | D |
Oral prednisolone is often used in combination with dapsone with very good effect. Complete remission can be achieved within months and children seem to respond much better than adults [11]. Other treatment options are usually not required, but include azathioprine, colchicine, mycophenolate mofetil, gold, and intravenous immunoglobulins (ivIG) [9].
Ectodermal Dysplasia with Skin Fragility (EDSF)
EDSF is a rare autosomal recessive genodermatosis affecting skin, nails, and hair. The international consensus for classification of epidermolysis bullosa (EB) [12] now considers EDSF to be a suprabasal form of EBS. It is due to loss of function mutations in the PKP1 gene that encodes for plakophilin, a protein that is a component of the desmosomal plaque.
It presents with:
Skin fragility (with flaccid blisters and erosions on minor trauma)
Hypotrichosis or alopecia
Focal palmoplantar keratoderma (with painful fissuring)
Hypohidrosis, nail dystrophy, and cheilitis may also be present [1]
Management Strategies
Management strategies include diagnosis and prevention and management of blisters similarly to EB.
Investigations
For diagnosis |
Skin biopsy for immunohistochemical antigenic mapping |
Skin biopsy for transmission electron microscopy (ultrastructural analysis) |
Skin biopsy for light microscopy |
Molecular testing |
Histological findings would show hyperkeratosis, acanthosis with widened intercellular spaces and acantholytic keratinocytes. Electron microscopy in EDSF demonstrates poorly developed, small desmosomes as well as a reduction in the number of desmosomes in the epidermis, particularly the lower suprabasal layer. Immunohistochemical analysis would reveal a complete absence of staining for plakophilin 1 [13].
Specific Therapies
Management of skin fragility is similar to that of epidermolysis bullosa, and includes careful handling of patients to prevent skin friction or trauma and managing blisters, as well as prevention and treatment of infections (see above, EB).
Kindler Syndrome
Kindler syndrome is a rare autosomal recessive skin fragility disorder characterized by trauma induced blistering in infancy, followed by photosensitivity and progressive poikiloderma on sun-exposed areas in later childhood [14]. Early diagnosis is difficult, as it may resemble dystrophic EB in childhood [15]. The abnormality is in the protein, kindlin-1 (fermitin family homologue 1) which is thought to be involved in connecting the actin cytoskeleton to the extracellular matrix. Loss of function mutations occur in the identified gene FERMT1 (formerly known as KIND1) located on chromosome 20 [16]. It is now thought to be part of EBS—mixed subtype [12].
Clinical Features
Blistering is usually acral, but more extensive blistering has also been reported.
Significant photosensitivity leading to skin atrophy and poikiloderma; this becomes more generalized and may involve non-sun exposed sites too [14].
Rarer presentations include: nail dystrophy, fusion of the digits, urethral, esophageal or anal stenosis, fusion of labia and chronic inflammation of the oral mucosa (dental caries, periodontitis, angular cheilitis, and desquamative gingivitis).
Squamous cell carcinoma of the lip and hard palate may also occur [15].
Management Strategies
Management strategies include prompt recognition and diagnosis. Prevention of blistering (similar to EB patients) and sun damage avoidance are very important. Long-term screening and monitoring for squamous cell carcinoma is recommended.
Investigations
Skin biopsy for immunohistochemical antigenic mapping |
Skin biopsy for transmission electron microscopy (ultrastructural analysis) |
Skin biopsy for light microscopy |
Genetic testing |
Light microscopy shows features of poikiloderma, which includes hyperkeratosis, epidermal atrophy, vacuolization of the basal layer, capillary dilation, and dermal edema. There would be cleavage at or close to the dermo-epidermal junction, near the basal keratinocyte layer, or beneath it. There may also be disruption of the collagen and elastic fibers in the papillary dermis.
Electron microscopy examination is used to differentiate from EB. Multiple planes of split may be seen within basal keratinocytes and/or in the lamina lucida, as well as reduplication of the lamina densa. The hemidesmosomes and anchoring fibrils and filaments are unaffected.
Immunohistochemical staining with anti-kindlin 1 antibody would show reduced or absent staining in the dermis [15].
Specific Therapies
Therapies in Kindler syndrome are similar to other subtypes of EB (see above). The blistering will improve with age.
In addition to these management strategies, sun protection advice (sun avoidance and use of sunscreen) should be given to delay the onset and severity of the poikiloderma. Regular skin checks should be performed by a dermatologist, due to the risk of developing squamous cell carcinomas [16].
Pemphigus
Pemphigus is a group of autoimmune intraepidermal vesiculobullous diseases that are characterized by the presence of antibodies to desmosomal proteins. It is classified into two main groups:
- 1.
Suprabasal type, which includes pemphigus vulgaris (PV) and pemphigus vegetans
- 2.
Superficial type, which includes pemphigus foliaceus (PF) and pemphigus erythematosus
Pemphigus is rare in children, infrequently seen before puberty. The most common form is PV, followed by PF. Pemphigus vegetans and erythematosus are exceedingly rare in children.
In PV, blisters develop on normal-appearing skin. They are flaccid and rupture easily, leaving painful erosions and crusts (Fig. 13.2). Scarring is unusual. Mucosal involvement, especially oral, is common and may precede skin lesions by several months (Fig. 13.3). Genital and ocular mucosal involvement occurs less frequently [9].
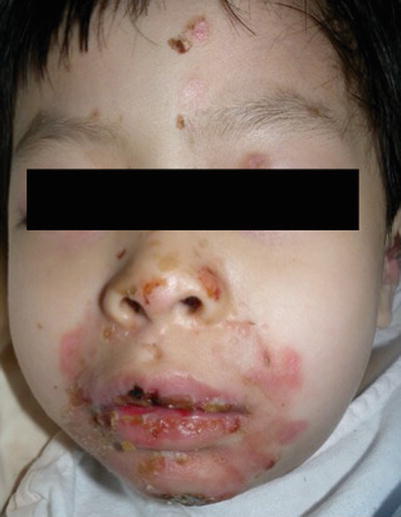
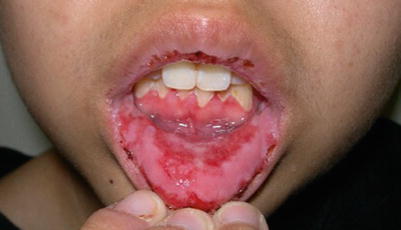
Fig. 13.2
Extensive blistering leading to erosions in pemphigus vulgaris
Fig. 13.3
Extensive erosions on the buccal mucosa in a teen with pemphigus vulgaris
There are two major forms of PF: an endemic form, Fogo selvegam, and a non-endemic form. The endemic form is most commonly seen in South America, especially in children who live in close proximity to rivers where the blackfly, Simulium nigrimanum, (thought to be the vector of this disease) is found. The etiology of the non-endemic form is unknown. The lesions usually present on the scalp, face, and upper trunk as erythematous scaly erosions. Blisters are usually too superficial to allow significant fluid accumulation. Mucosal involvement is minimal to none [14].
Investigations
For diagnosis |
Skin biopsy (lesional) for routine microscopy |
Skin biopsy (perilesional) for direct immunofluorescence (DIF) |
Serum for indirect immunofluorescence (IIF) |
Pemphigus Vulgaris [9]
Histology under routine light microscopy shows suprabasal acantholysis with basal keratinocytes remaining attached to the epidermal basement membrane. The dermal infiltrate as well as the blister cavity usually consists of lymphocytes, neutrophils, and eosinophils.
DIF shows deposition of IgG around keratinocytes, giving rise to a “crazy paving” pattern. IIF of the serum usually shows the presence of circulating IgG autoantibody; the titers correlate with clinical severity and can therefore be used to monitor progression. Antibodies are against desmoglein 3 when only mucosal lesions are present, and both desmogleins 1 and 3 in mucocutaneous disease.
Pemphigus Foliaceus/Erythematosus
Histology shows subtle acantholysis and occasionally subcorneal separation. A mild dermal lymphocytic and eosinophilic infiltrate may be found. DIF will usually show a linear deposition of IgG or IgM at the epidermal BMZ. The antigen in PF and erythematosus is desmoglein 1. The prognosis is good with very few fatalities.
Specific Therapies (For PV and PF)
Treatment for PF is similar to that for PV, but due to its less aggressive course, topical corticosteroids are usually sufficient [17].
Table 13.4
First line therapies
Modality | Dosage | Level of evidence |
|---|---|---|
Isolated, persistent lesions | ||
Topical or intralesional corticosteroids | BID | E |
Widespread disease | ||
Systemic corticosteroids (prednisolone) | 1–3 mg/kg/day | D |
A review article of all cases of childhood PV identified 47 cases from 20 studies of childhood PV [18]. The authors highlight the beneficial use of topical or intralesional corticosteroids for limited disease in removing debris, promoting healing, and offering symptomatic relief. It has been suggested that oral prednisolone be commenced at 1–1.5 mg/kg/day for a maximum of 3 months and, if the response is adequate, it can be switched to alternate-day therapy and gradually tapered and eventually discontinued. If the patient does not respond to this dosage, then this is an indication for using concomitant therapies (as listed below).
Table 13.5
Second line therapies
These are used for a steroid-sparing effect, and are often used as an adjuvant to oral corticosteroid therapy. | ||
Modality | Dosage | Level of evidence |
Azathioprine | 2 mg/kg/day divided into 2 doses then 1 mg/kg/day once a day as maintenance | D |
Dapsone | 50 mg–200 mg/day | E |
Cyclophosphamide | 50–150 mg/day | E |
Gold | 15 mg/week | E |
Methylprednisolone | 1–6 mg/kg/day | E |
Rituximab | 500 mg twice a day, 15 days apart or 375 mg/m2 body surface area twice a day, 15 days apart | D |
A review article describes 46 cases of childhood PV [19]. The mean age of onset of disease activity was 12 years. Systemic corticosteroids are the recommended mainstay of treatment, even in indolent cases, with an initial dose of 2–3 mg/kg/day with a slow tapering to 0.5–0.8 mg/kg/day in 2 weeks. Adjuvant treatments should be considered for steroid-sparing purposes. Azathioprine was the most commonly used adjuvant in seven cases. The authors recommend that approximately 2 mg/kg/day of azathioprine be used initially, divided into two doses, followed by a maintenance dose of 1 mg/kg once daily.
Another review article analysing 29 reports of PV showed that 26 patients had treatment with oral prednisolone either alone or with adjuvant therapy [20]. In those who had adjuvant therapy, six had azathioprine at 1–4 mg/kg/day, five had methylprednisolone at 1–6 mg/kg/day, four had dapsone at 14–200 mg/day, two were treated with cyclophosphamide, two with gold therapy, and one with cyclosporine. Patients who received complete or partial remission were those on oral prednisolone alone or in combination. Three patients responded well to a combination of methylprednisolone and azathioprine, two to rituximab infusions, and three to topical steroids.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


