Genetic Immunodeficiency Diseases
Primary immunodeficiency diseases are inherited disorders of the immune system that result in an increased susceptibility to infection and an increased morbidity and mortality. Many of these genetic immunodeficiency diseases may be associated with a variety of cutaneous abnormalities, and recognition of these clinical features may allow an early diagnosis of primary immunodeficiency. Cutaneous abnormalities may include cutaneous infections, atopic- or seborrheic-like dermatitis, macular erythemas, alopecia, poor wound healing, purpura, petechiae, telangiectasias, pigmentary dilution, cutaneous granulomas, extensive warts, angioedema, and lupus-like changes (Table 143-1). Other clinical features often include failure to thrive, visceral infection, autoimmune disorders, connective tissue/rheumatologic diseases, allergic reactions, and neoplasias.
Cutaneous Manifestations | Associated Immunodeficiency | Cutaneous Manifestations | AssociatedImmunodeficiency |
|---|---|---|---|
| Hereditary angioedema IgA deficiency/IgM deficiency Chronic granulomatous disease Common variable immunodeficiency Elevated IgM with hypogammaglobulinemia Ectodermal dysplasia with immunodeficiency Hyperimmunoglobulinemia E syndrome Severe combined immunodeficiency Wiskott–Aldrich syndrome X-linked agammaglobulinemia |
| Ataxia-telangiectasia |
| Chédiak–Higashi syndrome Griscelli syndrome Wiskott–Aldrich syndrome Chédiak–Higashi syndrome Griscelli syndrome IgA deficiency Chédiak–Higashi syndrome Chronic granulomatous disease Hyperimmunoglobulinemia E syndrome Leukocyte adhesion deficiency | ||
| Chronic granulomatous disease Hyperimmunoglobulinemia E syndrome Leukocyte adhesion deficiency |
| X-linked hypogammaglobulinemia Ataxia-telangiectasia “Leiner” phenotype: complement deficiency or dysfunction Severe combined immunodeficiency X-linked agammaglobulinemia Common variable immunodeficiency Elevated IgM with hypogammaglobulinemia Epidermodysplasia verruciformis (see Chapter 196) IgM deficiency Severe combined immunodeficiency after transplantation (mutations in IL2RG or JAK3) WHIM syndrome |
| Ataxia-telangiectasia Chronic granulomatous disease Chronic mucocutaneous candidiasis Common variable immunodeficiency Severe combined immunodeficiency, especially TAP2 deficiency X-linked hypogammaglobulinemia |
| |
| Chronic mucocutaneous candidiasis DiGeorge syndrome Hyperimmunoglobulinemia E syndrome Severe combined immunodeficiency especially TAP2 deficiency | ||
| Hyperimmunoglobulinemia E syndrome | ||
| DiGeorge syndrome Severe combined immunodeficiency | ||
| IgA deficiency Chronic granulomatous disease, autosomal recessive Chronic granulomatous disease, X-linked carrier Common variable immunodeficiency Complement deficiency, early components Elevated IgM with hypogammaglobulinemia |
Immunodeficiency should be suspected when patients have recurrent infections of increased duration or severity, particularly with unusual organisms. Incomplete clearing of infections, unexpected or severe complications of infection, or poor response to antibiotics may be associated.1 Affected infants often grow poorly (failure to thrive). The most common noncutaneous abnormalities are infections, diarrhea, vomiting, hepatosplenomegaly, arthritis, adenopathy or paucity of lymph nodes/tonsils, and hematologic abnormalities.
The classification of genetic immunodeficiency disorders includes (1) antibody deficiencies, (2) cellular deficiencies, (3) combined antibody and cellular deficiencies, (4) disorders of phagocytosis and cell killing, and (5) complement defects. The characteristic clinical signs of each group suggest that proper classification and laboratory tests may be used to confirm the diagnosis. The laboratory testing and clinical patterns of illness associated with each group of immunodeficiency disorders that allow their differential diagnosis are outlined in Table 143-2 and Fig. 143-1. The most important disorder in the differential diagnosis of all genetic immunodeficiency disorders is human immunodeficiency virus (HIV) infection. In addition to the lack of HIV antigen as detected by polymerase chain reaction in patients with genetic immunodeficiency, other features help to differentiate the disorders. Patients with HIV infection tend to show an inverted CD4/CD8 ratio and hypergammaglobulinemia, in contrast to the hypogammaglobulinemia of many patients with genetic immunodeficiency.
Disorder | Infection | Other |
|---|---|---|
Antibody | Sinopulmonary (pyogenic bacteria) Gastrointestinal (enterovirus, Giardia) Normal handling of fungal and viral infections (exception is enterovirus) | Autoimmune disease (autoantibodies, inflammatory bowel disease) Minimal growth retardation Paucity of lymphoid tissue |
Cellular | Low-grade or opportunistic infections Pneumonia (pyogenic bacteria, Pneumocystis, viruses) Gastrointestinal (viruses) Skin, mucous membranes (fungi) | Growth retardation Graft-versus-host disease Fatal infections from live vaccines Malignancy |
Phagocytosis and cell killing | Skin, reticuloendothelial system (Staphylococcus, enteric bacteria, Aspergillus, Mycobacteria) | Ulcerative stomatitis |
Complement | Alternative, late components Sepsis/blood-borne (Streptococci, Pneumococci, Neisseria) | Early components Autoimmune disease (systemic lupus erythematosus, glomerulonephritis) C1 esterase inhibitor deficiency Angioedema |
Figure 143-1
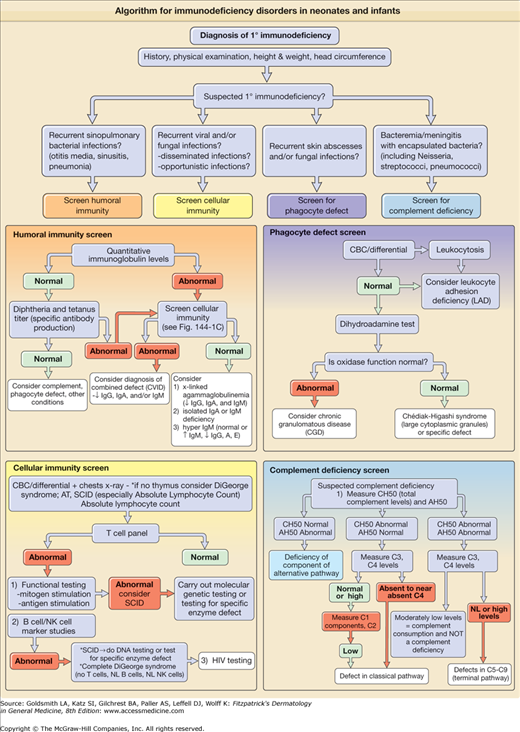
A–C. Algorithm for immunodeficiency disorders in neonates and infants. If the clinical presentation is concerning for severe combined immunodeficiency (SCID), this is a medical emergency and patient needs immediate referral to an immunologist. AT = ataxia-telangiectasia; CBC = complete blood cell count; CVID = common variable immunodeficiency; HIV = human immunodeficiency virus; Ig = immunoglobulin; NK cell = natural killer cell; NL = normal; ↓ = decreased; ↑ = increased.
Antibody Deficiency Disorders
|
Agammaglobulinemia results from gene defects that prevent the assembly of a full B cell antigen receptor. X-linked agammaglobulinemia (XLA) is the most common cause of agammaglobulinemia and results from defects in a cytoplasmic tyrosine kinase, Bruton’s tyrosine kinase (BTK). XLA is inherited in an X-linked fashion, and approximately 50% of affected boys have a family history of the disorder.2 Autosomal recessive agammaglobulinemia affects males and females equally and results from defects in the genes that encode for components of the pre-B cell and B cell receptors or in BLNK, a scaffold protein that assembles signaling molecules associated with the pre-B cell and B cell receptor.23
 The underlying defect in agammaglobulinemia is failure of maturation of a pre-B cell into an immature B cell; early B-cell precursors are found in the bone marrow in normal numbers. The causative genes, including BTK, participate in B-cell receptor signaling and are essential for B-cell maturation. Cell-mediated immunity is normal. More than 500 different mutations in BTK have been reported, and no single mutation is detected in more than 3% of the patients.4 Carrier detection in XLA is possible by analyzing the patterns of X-chromosome inactivation, with selective inactivation of the abnormal X chromosome in B lymphocytes from female carriers or by intracellular staining and flow cytometry for BTK in monocytes.
The underlying defect in agammaglobulinemia is failure of maturation of a pre-B cell into an immature B cell; early B-cell precursors are found in the bone marrow in normal numbers. The causative genes, including BTK, participate in B-cell receptor signaling and are essential for B-cell maturation. Cell-mediated immunity is normal. More than 500 different mutations in BTK have been reported, and no single mutation is detected in more than 3% of the patients.4 Carrier detection in XLA is possible by analyzing the patterns of X-chromosome inactivation, with selective inactivation of the abnormal X chromosome in B lymphocytes from female carriers or by intracellular staining and flow cytometry for BTK in monocytes.
Agammaglobulinemia is characterized by recurrent pyogenic infections that often begin between 3 and 18 months after birth, concurrent with the waning of maternal immunoglobulins (Ig). These patients have absent or barely detectable tonsils and cervical lymph nodes.5 Skin infections, especially furunculosis and impetigo, occur in 28% of patients and often surround body orifices. An atopic-like eczematous eruption that fails to improve with Ig therapy has been described in many affected children. Pyoderma gangrenosum and noninfectious cutaneous granulomas have been reported. Childhood exanthematous disorders are handled appropriately, but the infections may recur, owing to a failure to develop specific antibodies.
Recurrent otitis, sinusitis, bronchitis, and pneumonia are the earliest infectious manifestations and usually are caused by Pneumococci, Staphylococci, or Haemophilus. Untreated pulmonary infections may lead to progressive bronchiectasis and chronic pulmonary disease, seen in 45% of XLA patients over the age of 10.6 Patients can also suffer from chronic enteroviral infections and hearing loss from repeated otitis and sinusitis infections. Other common bacterial infections include conjunctivitis, osteomyelitis, septic arthritis, and meningitis. Protracted diarrhea may be due to infection, particularly with Giardia, Salmonella, Campylobacter, or Cryptosporidium spp. Three virus groups cause problems: (1) enterovirus, (2) hepatitis B virus, and (3) rotavirus. Patients have developed paralysis after administration of the live polio vaccine. A rheumatoid-like arthritis, characterized by chronic inflammation and swelling of the large joints, may develop in as many as one-third to one-half of boys with XLA and is often due to mycoplasmal infection (Ureaplasma urealyticum). Disseminated echovirus infection has caused meningoencephalitis and a dermatomyositis-like disorder with brawny edema, induration of the muscles with accompanying weakness, muscle contractures, and poikiloderma.
The diagnosis of agammaglobulinemia is made by serum concentrations of IgG, IgA, and IgM that are far below the 95% confidence limits for appropriate controls (usually less than 100 mg/dL total Ig) and by the virtual absence of B cells in the peripheral circulation (<1% of normal). Identification of a defect in one of the known genetic causes of agammaglobulinemia confirms the diagnosis and allows for genetic counseling and prenatal diagnosis.
Early Ig replacement, intravenously (IVIG) or subcutaneously, and antibiotic use markedly reduces the risk of infections, although it may not be helpful in diminishing the risk and morbidity of chronic lung disease or chronic enterovirus infection.
|
 The disease prevalence of common variable immunodeficiency (CVI) is estimated at 1 in 25,000 and affects males and females equally.7,8 The onset of symptoms occurs at any age, with male patients presenting earlier than female patients; the mean age of onset of symptoms is 23 and 28 years, and mean age of diagnosis of 29 and 33 years, respectively.9 A minimum age of 4 years is used to exclude patients with other primary immunodeficiency diseases.10 Most cases are sporadic, but at least 10% are familial with a predominance of autosomal dominant over autosomal recessive inheritance.
The disease prevalence of common variable immunodeficiency (CVI) is estimated at 1 in 25,000 and affects males and females equally.7,8 The onset of symptoms occurs at any age, with male patients presenting earlier than female patients; the mean age of onset of symptoms is 23 and 28 years, and mean age of diagnosis of 29 and 33 years, respectively.9 A minimum age of 4 years is used to exclude patients with other primary immunodeficiency diseases.10 Most cases are sporadic, but at least 10% are familial with a predominance of autosomal dominant over autosomal recessive inheritance.
 Identified genetic defects underlying CVI result in abnormalities in transmembrane activator, calcium modulator, and cyclophilin ligand interactor (TACI), inducible costimulator (ICOS), B-cell activation factor of the tumor necrosis factor (TNF) family receptor (BAFF-R), and CD19. Each of these is critical for B-lymphocyte activation and differentiation. Altogether, mutations in these genes account for approximately 10% to 20% of patients, with defects in TACI being the most common.7 The majority of defects are yet to be discovered.
Identified genetic defects underlying CVI result in abnormalities in transmembrane activator, calcium modulator, and cyclophilin ligand interactor (TACI), inducible costimulator (ICOS), B-cell activation factor of the tumor necrosis factor (TNF) family receptor (BAFF-R), and CD19. Each of these is critical for B-lymphocyte activation and differentiation. Altogether, mutations in these genes account for approximately 10% to 20% of patients, with defects in TACI being the most common.7 The majority of defects are yet to be discovered.
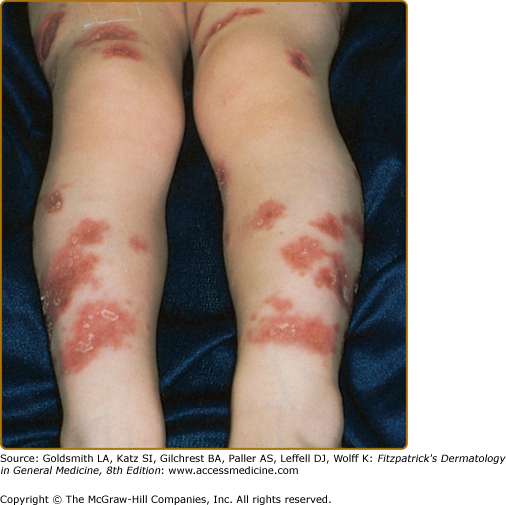
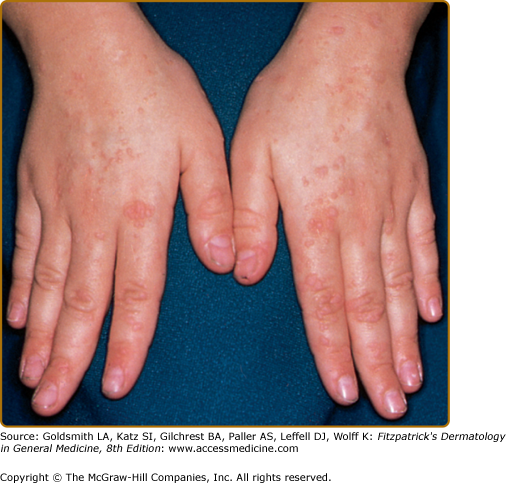
CVI commonly presents in young adults, but 25% of cases are diagnosed before the age of 21 years.9 Patients have infections similar to those in patients with XLA, particularly sinopulmonary infections, but are less susceptible to enteroviral infections and more susceptible to Giardia infections. Many patients with CVI have liver disease and gastrointestinal (GI) disease, causing malabsorption syndromes. Noncaseating granulomas of skin (Fig. 143-2), lungs, liver, and spleen have been reported. Caseating granulomas of the skin and viscera, although rare, have also been described.11,12 Extensive warts can be a major problem in individuals with CVI (Fig. 143-3). Lymphoid tissues often are enlarged, and splenomegaly with hypersplenism is found in 25% of patients. Autoimmune disorders are especially frequent (11% to 22%),9,13 particularly autoimmune thrombocytopenia, autoimmune hemolytic anemia, rheumatoid arthritis, sicca syndrome, and pernicious anemia. Alopecia areata and lupus also have been described. In 10% to 20% of patients, at least one family member is also immunodeficient, particularly with CVI or IgA deficiency.14 The incidence of lymphoreticular malignancy and gastric carcinoma are markedly increased, particularly in the fifth and sixth decades of life.15
 Failure in B-cell differentiation and resultant impaired secretion of immunoglobulins (Ig) leads to reduction in serum IgG and IgA and/or IgM by two standard deviations or more below the mean. The numbers of circulating B lymphocytes are usually normal but may be decreased or absent. Cellular immunity may be impaired.16 Approximately one-half of patients have T-cell dysfunction,17 with the incidence increasing with advancing age. Some patients have a decreased absolute number of circulating CD4+ T cells and a normal number of CD8+ T cells.18 The diagnosis of CVI requires the exclusion of known genetic causes of other immunodeficiency diseases and a 2-year period free of lymphoma.8
Failure in B-cell differentiation and resultant impaired secretion of immunoglobulins (Ig) leads to reduction in serum IgG and IgA and/or IgM by two standard deviations or more below the mean. The numbers of circulating B lymphocytes are usually normal but may be decreased or absent. Cellular immunity may be impaired.16 Approximately one-half of patients have T-cell dysfunction,17 with the incidence increasing with advancing age. Some patients have a decreased absolute number of circulating CD4+ T cells and a normal number of CD8+ T cells.18 The diagnosis of CVI requires the exclusion of known genetic causes of other immunodeficiency diseases and a 2-year period free of lymphoma.8
 Ig replacement is standard treatment for CVI. Prophylactic antibiotics should be initiated in patients who continue to have infections despite Ig therapy.19 Patients with granulomatous inflammation have a worse prognosis.20 The mean age of death for females is 45.5 years and for males 40 years. The majority of patients die from either lymphoma or chronic sinopulmonary infections.9
Ig replacement is standard treatment for CVI. Prophylactic antibiotics should be initiated in patients who continue to have infections despite Ig therapy.19 Patients with granulomatous inflammation have a worse prognosis.20 The mean age of death for females is 45.5 years and for males 40 years. The majority of patients die from either lymphoma or chronic sinopulmonary infections.9
Selective Immunoglobulin Disorders
|
 IgA deficiency is often sporadic, but both autosomal recessive and autosomal dominant forms of inheritance have been described. Susceptibility to IgA has been linked to the HLA-DQ/DR locus and an extended major histocompatibility complex (MHC) haplotype (HLA-B8, SC01, DR3) is found with increased frequency in both IgA deficiency and CVI.9,15,21 Mutations in the TNF receptor family member TACI are also found in patients with IgA deficiency, accounting for about 5% of patients. B cells from individuals with TACI mutations do not produce IgG and IgA in response to the TACI ligand, reflecting impaired isotype switching.7 IgA deficiency occurs in approximately 1 in 600 persons, and most of those affected are healthy. However, affected individuals tend to have an increased incidence of upper respiratory tract infections (especially viral), allergies, atopic dermatitis, chronic gastroenteritis, and autoimmune disorders with circulating autoimmune antibodies.22 Individuals prone to infection should be screened for functional defects. Patients with no detectable IgA who have the capacity to synthesize specific antibodies may develop IgE, anti-IgA antibodies with infusion of blood products that contain IgA,23 and risk transfusion reactions with subsequent blood product infusions including IVIG. However, the incidence of anti-IgA reactions in IVIG administration is rare and life-saving blood product infusions should not be withheld in an IgA-deficient patient, especially if it is the patient’s first infusion.
IgA deficiency is often sporadic, but both autosomal recessive and autosomal dominant forms of inheritance have been described. Susceptibility to IgA has been linked to the HLA-DQ/DR locus and an extended major histocompatibility complex (MHC) haplotype (HLA-B8, SC01, DR3) is found with increased frequency in both IgA deficiency and CVI.9,15,21 Mutations in the TNF receptor family member TACI are also found in patients with IgA deficiency, accounting for about 5% of patients. B cells from individuals with TACI mutations do not produce IgG and IgA in response to the TACI ligand, reflecting impaired isotype switching.7 IgA deficiency occurs in approximately 1 in 600 persons, and most of those affected are healthy. However, affected individuals tend to have an increased incidence of upper respiratory tract infections (especially viral), allergies, atopic dermatitis, chronic gastroenteritis, and autoimmune disorders with circulating autoimmune antibodies.22 Individuals prone to infection should be screened for functional defects. Patients with no detectable IgA who have the capacity to synthesize specific antibodies may develop IgE, anti-IgA antibodies with infusion of blood products that contain IgA,23 and risk transfusion reactions with subsequent blood product infusions including IVIG. However, the incidence of anti-IgA reactions in IVIG administration is rare and life-saving blood product infusions should not be withheld in an IgA-deficient patient, especially if it is the patient’s first infusion.

Cellular Deficiencies
 The incidence of X-linked lymphoproliferative disease (XLP) is 1 in 3 million males.24
The incidence of X-linked lymphoproliferative disease (XLP) is 1 in 3 million males.24
 XLP disease results from mutations in SH2D1A,25 which encodes an adapter protein, signaling lymphocytic activation molecule (SLAM)-associated protein (SAP), critical to intracellular signaling pathways.26 SAP is expressed in T cells, natural killer (NK) cells, and NKT cells. Patients have no NKT cells in the periphery and defective SAP-mediated activation of their NK and CD8+ T cells.27
XLP disease results from mutations in SH2D1A,25 which encodes an adapter protein, signaling lymphocytic activation molecule (SLAM)-associated protein (SAP), critical to intracellular signaling pathways.26 SAP is expressed in T cells, natural killer (NK) cells, and NKT cells. Patients have no NKT cells in the periphery and defective SAP-mediated activation of their NK and CD8+ T cells.27
 XLP is characterized by fulminant infectious mononucleosis, dysgammaglobulinemia, and lymphoproliferative disorders.26 Patients are also at risk for the development of autoimmune disorders. These clinical manifestations usually develop following Epstein–Barr virus (EBV) infection in boys who have previously had normal immunologic responses. With EBV infection, however, patients respond abnormally to the antigen and fail to develop EBV-specific serologic responses.
XLP is characterized by fulminant infectious mononucleosis, dysgammaglobulinemia, and lymphoproliferative disorders.26 Patients are also at risk for the development of autoimmune disorders. These clinical manifestations usually develop following Epstein–Barr virus (EBV) infection in boys who have previously had normal immunologic responses. With EBV infection, however, patients respond abnormally to the antigen and fail to develop EBV-specific serologic responses.
 Infectious mononucleosis, the most common clinical manifestation, affects 60% of patients and median age at onset is 3 years.26 Clinical signs include fever, pharyngitis, rash, lymphadenopathy, and hepatosplenomegaly. A progressive hypogammaglobulinemia is seen in 30% of patients before or after EBV infection; the median age of onset of hypogammaglobulinemia is 7–9 years.24,28 All patients with infectious mononucleosis and most patients with dysgammaglobulinemia have evidence of EBV infection. In contrast, the development of lymphomas can occur in XLP patients with no evidence of detectable EBV infection.29 Malignant lymphomas and nonmalignant lymphoproliferative disorders, including lymphomatoid granulomatosis, granulomatosis with polyangiitis (Wegener’s), and necrotizing vasculitis, affect 20% to 30% of patients.28 The majority of malignant tumors are of B-cell origin.
Infectious mononucleosis, the most common clinical manifestation, affects 60% of patients and median age at onset is 3 years.26 Clinical signs include fever, pharyngitis, rash, lymphadenopathy, and hepatosplenomegaly. A progressive hypogammaglobulinemia is seen in 30% of patients before or after EBV infection; the median age of onset of hypogammaglobulinemia is 7–9 years.24,28 All patients with infectious mononucleosis and most patients with dysgammaglobulinemia have evidence of EBV infection. In contrast, the development of lymphomas can occur in XLP patients with no evidence of detectable EBV infection.29 Malignant lymphomas and nonmalignant lymphoproliferative disorders, including lymphomatoid granulomatosis, granulomatosis with polyangiitis (Wegener’s), and necrotizing vasculitis, affect 20% to 30% of patients.28 The majority of malignant tumors are of B-cell origin.
 XLP is a fatal disease and 70% of patients die by the age of 10 years.27 The median survival after development of primary EBV infection and fulminant mononucleosis is 1–2 months.24 Most patients die from severe hepatitis, liver necrosis, and hepatic failure. Patients with isolated hypogammaglobulinemia treated with Ig replacement have a better prognosis than those who develop fulminant infectious mononucleosis or lymphoma.26
XLP is a fatal disease and 70% of patients die by the age of 10 years.27 The median survival after development of primary EBV infection and fulminant mononucleosis is 1–2 months.24 Most patients die from severe hepatitis, liver necrosis, and hepatic failure. Patients with isolated hypogammaglobulinemia treated with Ig replacement have a better prognosis than those who develop fulminant infectious mononucleosis or lymphoma.26
|
Several clinical subtypes of chronic mucocutaneous candidiasis (CMC) have been defined (Table 143-3). They have varied clinical manifestations, variable immunodeficiency, and different forms of genetic inheritance. Patients with CMC may have childhood or mature onset, familial, or sporadic occurrence, and CMC may be present with or without endocrinopathy. Patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED or autoimmune polyendocrine syndrome or APS, type 1) often have affected siblings. APECED and other familial forms of CMC are autosomal recessive. Autosomal dominant inheritance is seen in patients with associated keratitis.
CMC Type | Inheritance/Gene Defect | Onset | Clinical Features | Associated Disorders | NonCandidal Infections |
|---|---|---|---|---|---|
Chronic oral candidiasis | No known genetic defect | Middle-aged or elderly women | Candidiasis of tongue and buccal mucosa No esophageal, skin, or nail involvement | Fe2+ deficiency | No |
Familial chronic mucocutaneous candidiasis | Autosomal recessive and autosomal dominant Males and females are equally affected | Early childhood, often before age 2 years | Oral candidiasis Limited skin and nail involvement | No endocrinopathies | Yes |
Autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy syndrome | Autosomal recessive Mutations in the AIRE (autoimmune regulator) gene on 21q22.3 More common in the Finnish population, Iranian Jews, and Sardinians Rarely, dominant form with AIRE mutations or mapped to chromosome 2p | Candidal infections before age 5 years Endocrine abnormalities between 10 and 15 years of age | Oral and diaper area candidiasis more often than skin and nail involvement Endocrinopathies and autoimmune disorders | Most common endocrinopathies: Hypoparathyroidism Hypoadrenalism Other associated disorders: Thyroid disease Primary hypogonadism Hepatitis Malabsorption Pernicious anemia Alopecia areata Vitiligo Ectodermal manifestations: Dental enamel hypoplasia Pitted nail dystrophy Keratoconjunctivitis Myasthenia gravis Hypogammaglobulinemia | Yes |
CMC with thymoma | No known genetic basis | Adult onset | Mucous membrane and cutaneous candidiasis | Malignant/benign thymomas Aplastic anemia Myasthenia gravis Hypogammaglobulinemia | No |
Chronic localized candidiasis (candida granuloma) | No known genetic basis Males and females are equally affected | Early childhood, often before age 5 years | Thick, adherent candidal crusts on the scalp and face Oral candidiasis | None | Yes |
CMC with keratitis | Autosomal dominant | Early childhood | Candidiasis of the oral cavity, diaper area | Keratoconjunctivitis Alopecia Endocrine abnormalities | Yes |
 The clinical features of CMC may be seen in a variety of immunologic disorders, all characterized by ineffective defense mechanisms against Candida. In general, the patients with greater severity and an earlier onset of cutaneous candidal infections have more severe immunologic alterations. CMC patients have shown general dysregulation of interleukin 12 (IL-12), IL-6, and interferon-γ (IFN-γ) production,30 as well as autoantibodies to IL-17 and IL-2231 resulting in an inability to mount a cell-mediated response to clear candidal organisms. Chronic infections result in production of high levels of inflammatory cytokines (IL-6) followed by anti-inflammatory cytokines (IL-10) that further reduce the production of T helper 1 (Th1)-inducing cytokines via a positive feedback loop.30
The clinical features of CMC may be seen in a variety of immunologic disorders, all characterized by ineffective defense mechanisms against Candida. In general, the patients with greater severity and an earlier onset of cutaneous candidal infections have more severe immunologic alterations. CMC patients have shown general dysregulation of interleukin 12 (IL-12), IL-6, and interferon-γ (IFN-γ) production,30 as well as autoantibodies to IL-17 and IL-2231 resulting in an inability to mount a cell-mediated response to clear candidal organisms. Chronic infections result in production of high levels of inflammatory cytokines (IL-6) followed by anti-inflammatory cytokines (IL-10) that further reduce the production of T helper 1 (Th1)-inducing cytokines via a positive feedback loop.30
 Humoral immunity appears normal in most patients and 25% to 35% of patients with CMC have no demonstrable immunologic defects. Many patients with CMC have associated APECED syndrome,32 owing to mutations in the autoimmune regulator (AIRE) gene which maps to 21q22.3 and encodes a DNA transcription factor.33 Mice that are deficient in AIRE do not delete organ-specific T cells in the thymus, thus promoting the development of autoimmune disease.34 The reason for susceptibility to mucocutaneous candidal infections is unclear, but may be related to the decreased Th17 and Th1 immune responses.31 APECED must be distinguished from immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome, an X-linked recessive disorder in which the abnormal development of regulatory T cells results from mutations in the FOXP3 gene. Patients with IPEX syndrome show atopic or psoriasiform dermatitis, nail dystrophy, autoimmune endocrinopathies (overlapping with those seen in APECED syndrome), and autoimmune skin conditions such as alopecia universalis.35,36
Humoral immunity appears normal in most patients and 25% to 35% of patients with CMC have no demonstrable immunologic defects. Many patients with CMC have associated APECED syndrome,32 owing to mutations in the autoimmune regulator (AIRE) gene which maps to 21q22.3 and encodes a DNA transcription factor.33 Mice that are deficient in AIRE do not delete organ-specific T cells in the thymus, thus promoting the development of autoimmune disease.34 The reason for susceptibility to mucocutaneous candidal infections is unclear, but may be related to the decreased Th17 and Th1 immune responses.31 APECED must be distinguished from immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome, an X-linked recessive disorder in which the abnormal development of regulatory T cells results from mutations in the FOXP3 gene. Patients with IPEX syndrome show atopic or psoriasiform dermatitis, nail dystrophy, autoimmune endocrinopathies (overlapping with those seen in APECED syndrome), and autoimmune skin conditions such as alopecia universalis.35,36
Patients with CMC have recurrent, progressive infections of the skin, nails, and mucous membranes most commonly due to Candida albicans.33,37 Depending on the subtype, the clinical presentation ranges from recurrent, recalcitrant thrush (Fig. 143-4) to mild erythematous scaling plaques (see eFig. 143-4.1) with a few dystrophic nails to severe generalized, crusted granulomatous plaques (Fig. 143-5). The cutaneous plaques occur most commonly in intertriginous areas, periorificial sites, and the scalp, but they may be generalized. The nails are thickened, brittle, and discolored, and the paronychial areas are often erythematous, swollen, and tender. Scalp infections may lead to scarring and alopecia. Although the oral mucosa is the most frequent site of mucosal alteration, esophageal, genital, and laryngeal mucosae may be affected. Strictures may be formed by candidal infection at these mucosal sites. Scrapings and cultures from cutaneous or mucosal lesions demonstrate candidal organisms.
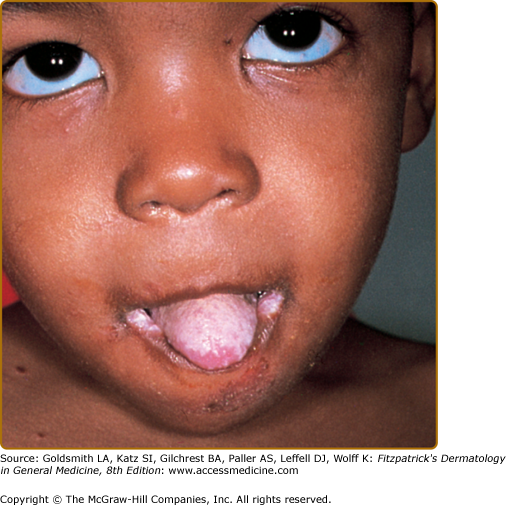
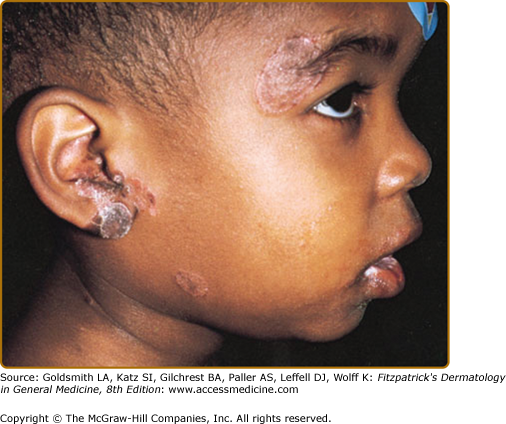
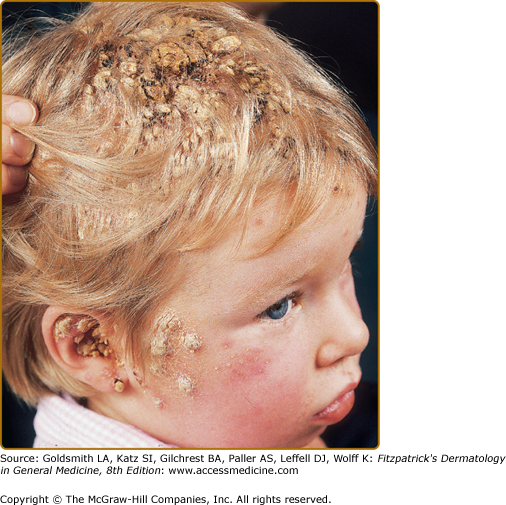
Patients with CMC rarely develop systemic candidiasis, but 50% may develop recurrent or severe infections due to other organisms. In one study, 81% of patients with early-onset CMC also had infections with bacteria, fungi, and parasites, including bacterial septicemia.33 Concomitant dermatophyte infections may occur.
In patients with APECED, the candidal infections tend to begin by 5 years of age, although the endocrinologic dysfunction may not be apparent until 12–13 years of age (see Fig. 143-5). The most commonly associated endocrinopathies are hypoparathyroidism (88%) and hypoadrenocorticism (60%). One-third of patients have candidiasis, hypoparathyroidism, and defective adrenal function. Other associated endocrinopathies or autoimmune disorders include gonadal insufficiency (45%), alopecia areata (20%), pernicious anemia (16%), thyroid abnormalities (12%), chronic active hepatitis or juvenile cirrhosis (9%), vitiligo, diabetes mellitus, and hypopituitarism. Chronic diarrhea and malabsorption have been reported in 25% of patients and usually are associated with hypoparathyroidism. Some affected patients also have pulmonary fibrosis, dental enamel hypoplasia, and keratoconjunctivitis. The “ectodermal dysplasia” features are likely to be secondary to the candidal infections or autoimmunity.37 Patients with APECED often have autoimmune antibodies, including antithyroglobulin, antimicrosomal, antiadrenal, and antimelanocyte antibodies, and rheumatoid factor. Autoantibodies also have been found in patients with CMC who do not have clinical endocrinologic disease.
Candidal lesions in patients with CMC generally respond to systemically administered azole antifungal agents (itraconazole, fluconazole) or terbinafine.38 Ketoconazole is no longer used because of the risk of hepatitis. Patients who are resistant usually respond to amphotericin B with or without flucytosine. Cutaneous granulomas often are less responsive despite clearance of infection. Recurrences are common, and the antifungal agents must be used intermittently. The drugs have no effect on the abnormal cell-mediated immunity. All patients with CMC should have an annual endocrine evaluation and patients with documented endocrinopathy or a family history of APECED should be monitored more closely. Patients who have a history of infections other than candidal should have further evaluation of their immune status.
 The incidence of DiGeorge syndrome (DGS) is 1 in 4,000 live births39 Ninety percent of the cases are associated with a deletion in chromosome 22qll.l.
The incidence of DiGeorge syndrome (DGS) is 1 in 4,000 live births39 Ninety percent of the cases are associated with a deletion in chromosome 22qll.l.
 DGS (congenital thymic aplasia) is a member of a group of disorders that result from deletion of chromosome 22q11 (CATCH 22/DGS/velocardiofacial syndrome).40 The disorder results from developmental defects of the third and fourth pharyngeal pouches due to haploinsufficiency of Tbx1, a t-box transcription factor.41 Five percent to 10% of patients do not have this deletion; some have deletion of chromosome 10 and others have no identifiable gene defect.42 All patients have T cell defects, but patients with partial DGS have only mild T cell abnormalities, showing an increase in T cell numbers from birth to age 2 that subsequently do not decrease with time.43 Patients with complete DGS (complete lack of thymus and T cell percentage less than 1 to 2) have SCID with B cell immunodeficiency as well, presumably due to the lack of T cell help for B cells. These patients do not recover functional T-cells throughout early infancy. Complete DGS occurs in less than 1% of cases.44 It is often found in conjunction with 22q11 hemizygosity,45,46 the CHARGE association,47,48 or diabetic embryopathy.49
DGS (congenital thymic aplasia) is a member of a group of disorders that result from deletion of chromosome 22q11 (CATCH 22/DGS/velocardiofacial syndrome).40 The disorder results from developmental defects of the third and fourth pharyngeal pouches due to haploinsufficiency of Tbx1, a t-box transcription factor.41 Five percent to 10% of patients do not have this deletion; some have deletion of chromosome 10 and others have no identifiable gene defect.42 All patients have T cell defects, but patients with partial DGS have only mild T cell abnormalities, showing an increase in T cell numbers from birth to age 2 that subsequently do not decrease with time.43 Patients with complete DGS (complete lack of thymus and T cell percentage less than 1 to 2) have SCID with B cell immunodeficiency as well, presumably due to the lack of T cell help for B cells. These patients do not recover functional T-cells throughout early infancy. Complete DGS occurs in less than 1% of cases.44 It is often found in conjunction with 22q11 hemizygosity,45,46 the CHARGE association,47,48 or diabetic embryopathy.49
 The thymic shadow is absent or reduced at birth. Infants often have neonatal tetany with hypocalcemia due to the aplastic parathyroid glands. The cardiac anomalies are most commonly truncus arteriosus, septal defects, and abnormal aortic arch vessels. Characteristic facial features of DGS include a short philtrum, low-set malformed ears, and hypertelorism.
The thymic shadow is absent or reduced at birth. Infants often have neonatal tetany with hypocalcemia due to the aplastic parathyroid glands. The cardiac anomalies are most commonly truncus arteriosus, septal defects, and abnormal aortic arch vessels. Characteristic facial features of DGS include a short philtrum, low-set malformed ears, and hypertelorism.
 Many patients have recurrent mucocutaneous candidal infections as neonates, as well as increased susceptibility to viral infections, Pneumocystis jiroveci, and other fungal infections. Graft-versus-host disease (GVHD) may develop in infants given nonirradiated blood products. A small percentage of patients have complete athymia and about one-third of them develop an eczematous dermatitis with lymphadenopathy driven by oligoclonal T cells, known as atypical complete DiGeorge anomaly. There is an overall increase of malignancy in DGS, particularly hepatoblastoma; in a cohort of patients under the age of 14, the overall risk of malignancy was 900 per 100,000, whereas the overall risk of malignancy in children under 14 years is 3.4 per 100,000.50
Many patients have recurrent mucocutaneous candidal infections as neonates, as well as increased susceptibility to viral infections, Pneumocystis jiroveci, and other fungal infections. Graft-versus-host disease (GVHD) may develop in infants given nonirradiated blood products. A small percentage of patients have complete athymia and about one-third of them develop an eczematous dermatitis with lymphadenopathy driven by oligoclonal T cells, known as atypical complete DiGeorge anomaly. There is an overall increase of malignancy in DGS, particularly hepatoblastoma; in a cohort of patients under the age of 14, the overall risk of malignancy was 900 per 100,000, whereas the overall risk of malignancy in children under 14 years is 3.4 per 100,000.50
 Patients with complete DGS usually die within the first 2 years of life.51 In rare cases, transplant of HLA-matched bone marrow or peripheral blood mononuclear cells restores T-cell function.52 Postnatal thymus transplant can restore T-cell function in patients with complete DGS.44,53
Patients with complete DGS usually die within the first 2 years of life.51 In rare cases, transplant of HLA-matched bone marrow or peripheral blood mononuclear cells restores T-cell function.52 Postnatal thymus transplant can restore T-cell function in patients with complete DGS.44,53
Cartilage–Hair Hypoplasia Syndrome
|
 Cartilage–hair hypoplasia (CHH) syndrome is an autosomal recessive disorder that is most common in Amish and Finnish individuals.54 There is a 4:1 female–male ratio.55
Cartilage–hair hypoplasia (CHH) syndrome is an autosomal recessive disorder that is most common in Amish and Finnish individuals.54 There is a 4:1 female–male ratio.55
 The disorder results from mutations in RMRP, the RNA component of a ribonucleoprotein endoribonuclease.56 RNase MRP cleaves RNA primers responsible for DNA replication in mitochondria and in the nucleolus processes pre-rRNA. CHH has been mapped to 9p13.57 Forty different mutations have been described, most commonly 70A>G. Mutations alter ribosomal processing, leading to altered cytokine signaling and cell cycle progression in terminally differentiating lymphocytes and chondrocytes.58 Most patients have defective cell-mediated immunity, and patients may be particularly susceptible to severe disseminated varicella.55 Fifty-seven percent of patients have a decreased CD4+ cell count with a decreased total count of T lymphocytes and a subnormal CD4+/CD8+ ratio.55 A subset of patients, particularly those of Finnish origin, also has defective humoral immunity; 35% of patients have a deficiency of IgA or IgG subclasses or a combination.55
The disorder results from mutations in RMRP, the RNA component of a ribonucleoprotein endoribonuclease.56 RNase MRP cleaves RNA primers responsible for DNA replication in mitochondria and in the nucleolus processes pre-rRNA. CHH has been mapped to 9p13.57 Forty different mutations have been described, most commonly 70A>G. Mutations alter ribosomal processing, leading to altered cytokine signaling and cell cycle progression in terminally differentiating lymphocytes and chondrocytes.58 Most patients have defective cell-mediated immunity, and patients may be particularly susceptible to severe disseminated varicella.55 Fifty-seven percent of patients have a decreased CD4+ cell count with a decreased total count of T lymphocytes and a subnormal CD4+/CD8+ ratio.55 A subset of patients, particularly those of Finnish origin, also has defective humoral immunity; 35% of patients have a deficiency of IgA or IgG subclasses or a combination.55
 Patients have fine, sparse, hypopigmented hair (eFig. 143-5.1), and metaphyseal dysostosis that results in short-limbed dwarfism. Patients may have soft, doughy skin with degenerated elastic tissue. Chronic oral fungal or viral infections as well as recurrent upper respiratory infections, otitis media, and pneumonias are related to defective cellular and humoral immunity. Associated pleiotropic features include Hirschsprung disease, deficient erythrogenesis, and an increased risk of malignancies,59 particularly non-Hodgkin lymphoma and basal cell carcinomas. There is a marked variation in the clinical phenotype of patients with RMRP mutations including severe immunodeficiency without any other features.60
Patients have fine, sparse, hypopigmented hair (eFig. 143-5.1), and metaphyseal dysostosis that results in short-limbed dwarfism. Patients may have soft, doughy skin with degenerated elastic tissue. Chronic oral fungal or viral infections as well as recurrent upper respiratory infections, otitis media, and pneumonias are related to defective cellular and humoral immunity. Associated pleiotropic features include Hirschsprung disease, deficient erythrogenesis, and an increased risk of malignancies,59 particularly non-Hodgkin lymphoma and basal cell carcinomas. There is a marked variation in the clinical phenotype of patients with RMRP mutations including severe immunodeficiency without any other features.60
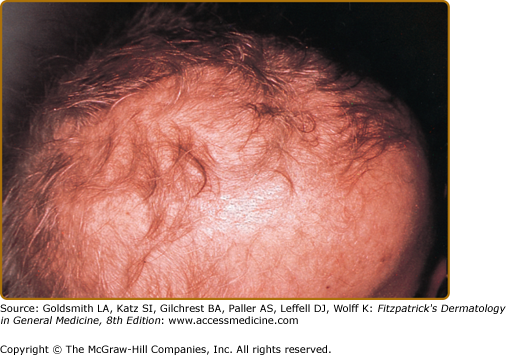
 Supportive therapy with appropriate antibiotic treatment is indicated. Bone marrow transplantation, although rarely performed, has fully corrected the immune deficiency but has no influence on the course of the chondroplasia or elastic tissue. A DNA marker-based analysis provides a useful method for prenatal diagnosis.
Supportive therapy with appropriate antibiotic treatment is indicated. Bone marrow transplantation, although rarely performed, has fully corrected the immune deficiency but has no influence on the course of the chondroplasia or elastic tissue. A DNA marker-based analysis provides a useful method for prenatal diagnosis.
Combined Antibody and T-Cell Deficiency
|
 The disorder usually occurs in males, with X-linked inheritance in approximately 70% of affected individuals.61 Incidence is approximately 1 in 1 million live births.62 An X-linked recessive form caused by mutations in CD40 ligand (CD154) gene and three autosomal recessive forms caused by mutations in CD40 or downstream signaling pathway components [activation-induced cytidine deaminase (AICD) and uracil-N-glycosylase (UNG)] have been described.63
The disorder usually occurs in males, with X-linked inheritance in approximately 70% of affected individuals.61 Incidence is approximately 1 in 1 million live births.62 An X-linked recessive form caused by mutations in CD40 ligand (CD154) gene and three autosomal recessive forms caused by mutations in CD40 or downstream signaling pathway components [activation-induced cytidine deaminase (AICD) and uracil-N-glycosylase (UNG)] have been described.63
 Hyper-IgM syndrome is caused by a defect of B-cell differentiation secondary to a failure of T–B cell interaction via CD40 ligand-CD40 pathway.62,63 Mature B cells expressing IgM and IgD on their surface develop normally but fail to undergo T cell (CD40 ligand)-dependent Ig isotype switching to produce IgG, IgA, or IgE antibodies. Some patients have detectable IgA levels in their serum, which is thought to occur in a CD40 ligand-independent manner. Ig isotype switching is a mechanism by which the immune system produces antibodies with different effector function while retaining variable region (antigen) specificity (see Chapter 10).
Hyper-IgM syndrome is caused by a defect of B-cell differentiation secondary to a failure of T–B cell interaction via CD40 ligand-CD40 pathway.62,63 Mature B cells expressing IgM and IgD on their surface develop normally but fail to undergo T cell (CD40 ligand)-dependent Ig isotype switching to produce IgG, IgA, or IgE antibodies. Some patients have detectable IgA levels in their serum, which is thought to occur in a CD40 ligand-independent manner. Ig isotype switching is a mechanism by which the immune system produces antibodies with different effector function while retaining variable region (antigen) specificity (see Chapter 10).
 Normally, isotype switching in B cells requires a contact-dependent signal from T cells delivered by CD40 ligand on activated T cells to CD40, a glycoprotein on the surface of B cells. Most patients with hyper-IgM syndrome have a mutation in the gene that encodes CD40 ligand, located on the X chromosome. T cells from patients with the syndrome cannot synthesize CD40 ligand, or in some cases a nonfunctional ligand is produced. B cells respond to antigen and produce specific antibodies; however, they are restricted to the IgM isotype, and there is no memory response. Patients with CD40 ligand deficiency may have defective antigen-induced T-cell proliferation64 and defective T-cell effector function.65, Elevations of IgM in the face of immunodeficiency have also been described in some patients with hypohidrotic ectodermal dysplasia due to defects in the gene encoding nuclear factor κB essential modulator (NEMO), also an X-linked recessive disease (see “Ectodermal Dysplasia with Immunodeficiency”).66–68
Normally, isotype switching in B cells requires a contact-dependent signal from T cells delivered by CD40 ligand on activated T cells to CD40, a glycoprotein on the surface of B cells. Most patients with hyper-IgM syndrome have a mutation in the gene that encodes CD40 ligand, located on the X chromosome. T cells from patients with the syndrome cannot synthesize CD40 ligand, or in some cases a nonfunctional ligand is produced. B cells respond to antigen and produce specific antibodies; however, they are restricted to the IgM isotype, and there is no memory response. Patients with CD40 ligand deficiency may have defective antigen-induced T-cell proliferation64 and defective T-cell effector function.65, Elevations of IgM in the face of immunodeficiency have also been described in some patients with hypohidrotic ectodermal dysplasia due to defects in the gene encoding nuclear factor κB essential modulator (NEMO), also an X-linked recessive disease (see “Ectodermal Dysplasia with Immunodeficiency”).66–68
 In contrast to X-linked agammaglobulinemia, female carriers of the hyper-IgM syndrome due to mutations in CD40 ligand have random inactivation of the X chromosome in T lymphocytes because CD40 ligand is not required for the normal development of T lymphocytes. Female carriers, in general, do not show clinical manifestations. However, a female carrier who was noted to have 95% of her T cells expressing the mutant X chromosome had recurrent infections of the upper and lower respiratory tract69 and another carrier had gastric lymphoma.70
In contrast to X-linked agammaglobulinemia, female carriers of the hyper-IgM syndrome due to mutations in CD40 ligand have random inactivation of the X chromosome in T lymphocytes because CD40 ligand is not required for the normal development of T lymphocytes. Female carriers, in general, do not show clinical manifestations. However, a female carrier who was noted to have 95% of her T cells expressing the mutant X chromosome had recurrent infections of the upper and lower respiratory tract69 and another carrier had gastric lymphoma.70
 More than one-half of patients develop symptoms of immunodeficiency and are diagnosed before age 1 year. Nearly all patients have symptoms by 4 years of age.62 Most notable are respiratory tract infections, seen in 81% of patients,62 dermatitis, an increased incidence and severity of warts, and oral ulcerations, sometimes in association with neutropenia.71,72 Cellulitis and subcutaneous abscesses are seen in 13% of patients.62 Recurrent diarrhea, central nervous system (CNS) infections, and sepsis are also common. Patients with X-linked hyper-IgM (CD40 ligand defects) and autosomal recessive defects in CD40 have a T cell immunodeficiency in addition to the antibody deficiency and are prone to opportunistic infections, including P. jiroveci.63 These patients have small lymph nodes without germinal centers. In contrast, patients with hyper-IgM syndrome caused by mutations in AICD or UNG have lymphoid hyperplasia, but do not appear to be susceptible to opportunistic infections because the defect only affects B cells and CD40-dependent costimulation of T cells by antigen-presenting cells is not affected. Hyper-IgM patients have an increased frequency of autoimmune disorders, especially of the hematopoietic system. Uncontrolled proliferation of IgM-producing plasma cells often occurs during the second decade of life, at times resulting in potentially fatal, massive infiltration of the GI tract, liver, and gallbladder. Patients with hyper-IgM syndrome also have an increased risk of cancer involving the GI tract. The sera of patients with hyper-IgM syndrome have very low amounts of IgG, IgA, and IgE. Although levels of IgM and IgD may be normal or high, high levels of IgM are actually found in fewer than one-third of patients.62
More than one-half of patients develop symptoms of immunodeficiency and are diagnosed before age 1 year. Nearly all patients have symptoms by 4 years of age.62 Most notable are respiratory tract infections, seen in 81% of patients,62 dermatitis, an increased incidence and severity of warts, and oral ulcerations, sometimes in association with neutropenia.71,72 Cellulitis and subcutaneous abscesses are seen in 13% of patients.62 Recurrent diarrhea, central nervous system (CNS) infections, and sepsis are also common. Patients with X-linked hyper-IgM (CD40 ligand defects) and autosomal recessive defects in CD40 have a T cell immunodeficiency in addition to the antibody deficiency and are prone to opportunistic infections, including P. jiroveci.63 These patients have small lymph nodes without germinal centers. In contrast, patients with hyper-IgM syndrome caused by mutations in AICD or UNG have lymphoid hyperplasia, but do not appear to be susceptible to opportunistic infections because the defect only affects B cells and CD40-dependent costimulation of T cells by antigen-presenting cells is not affected. Hyper-IgM patients have an increased frequency of autoimmune disorders, especially of the hematopoietic system. Uncontrolled proliferation of IgM-producing plasma cells often occurs during the second decade of life, at times resulting in potentially fatal, massive infiltration of the GI tract, liver, and gallbladder. Patients with hyper-IgM syndrome also have an increased risk of cancer involving the GI tract. The sera of patients with hyper-IgM syndrome have very low amounts of IgG, IgA, and IgE. Although levels of IgM and IgD may be normal or high, high levels of IgM are actually found in fewer than one-third of patients.62
 The leading causes of death are pneumonia, encephalitis, and malignancy. The condition is treated prophylactically with Ig replacement and P. jiroveci prophylaxis; neutropenia may respond to granulocyte-macrophage colony-stimulating factor. Few patients survive beyond the third decade.62 Allogeneic bone marrow transplantation can correct the immunodeficiency even if liver disease is present.73,74
The leading causes of death are pneumonia, encephalitis, and malignancy. The condition is treated prophylactically with Ig replacement and P. jiroveci prophylaxis; neutropenia may respond to granulocyte-macrophage colony-stimulating factor. Few patients survive beyond the third decade.62 Allogeneic bone marrow transplantation can correct the immunodeficiency even if liver disease is present.73,74
|
Wiskott–Aldrich syndrome (WAS) is an X-linked recessive disorder with an incidence of approximately 4 per million male births.75
The defective gene is WASP, mapped to Xp11.22–11.23, which encodes WASp, a hematopoietic specific cytoplasmic protein that functions in signaling and cytoskeletal organization. WASp couples signals arising at the cell membrane with reorganization of the cellular cytoskeleton, resulting in cellular activation and promotion of cell motility. Mutations in WASp affect organization of the immunologic synapse and T-cell activation, T and B lymphocyte migration, and initiation of the primary antibody response.76 There is a strong phenotype–genotype correlation; classic WAS occurs when WASp is absent or truncated, while X-linked thrombocytopenia occurs when mutated WASp is expressed.77 The atopic dermatitis is likely associated with the observed skewing of CD4+ T cell differentiation towards Th2 cells with suppression of Th1 and regulatory T cells (Treg) differentiation.78 Given the expression of WASp on epidermal Langerhans cells, abnormal interactions of Langerhans cells with T cells and ability of Langerhans cells to move to the lymph node after antigen stimulation may be involved as well.
WAS patients have decreased function and number of both T- and B-lymphocytes, beginning in the first years of life.79 The lymphocytes of patients with WAS lack microvilli formed by actin bundles, resulting in defective chemotaxis and, in some patients, there is decreased expression of sialoglycoproteins (e.g., CD43 and others) on lymphocytes and platelets. Defects in humoral immunity include abnormal serum Ig and decreased antibody response to polysaccharide antigens. WAS patients also have defects in NK cell cytotoxicity, dendritic cell migration, and activation, and impaired macrophage chemotaxis.76
The classic triad of WAS is (1) hemorrhage due to thrombocytopenia and platelet dysfunction, (2) recurrent pyogenic infections, and (3) recalcitrant dermatitis, but this triad appears in only 25% of patients.80 The bleeding diathesis is the most common manifestation of mutations in WAS, present in 84% of patients79 and often manifests initially during the first weeks or months of life with bloody diarrhea. Epistaxis, hematemesis, hematuria, mucocutaneous petechiae, and intracranial hemorrhage also may occur. Recurrent bacterial infections begin in infancy as levels of placentally transmitted maternal antibodies diminish. These infections include furunculosis, conjunctivitis, otitis media and otitis externa, pansinusitis, pneumonia, meningitis, and septicemia. Infections with encapsulated bacteria such as Pneumococcus, Haemophilus influenzae, and Neisseria meningitidis predominate. Patients are also susceptible to infections due to herpes and other viruses and to Pneumocystis jiroveci.
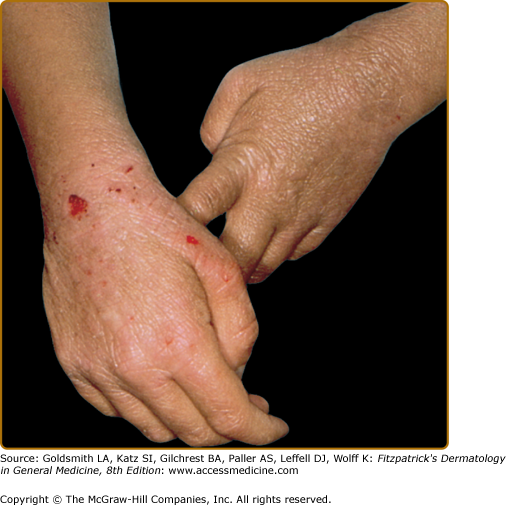
The atopic dermatitis associated with WAS, which occurs in approximately 80% of patients,75 usually develops during the first few months of life and may be quite severe. The face, scalp, and flexural areas are the most severely involved, although patients commonly have widespread involvement with progressive lichenification. The eruption may be more exfoliative than that of atopic dermatitis in individuals without WAS, and excoriated areas frequently have serosanguineous crusts (Fig. 143-6). Secondary bacterial infection of eczematous lesions is common, as are eczema herpeticum (see eFig. 143-6.1) and molluscum contagiosum. IgE-mediated allergic problems, such as urticaria, food allergies, and asthma, are seen in addition to the atopic dermatitis.
eFigure 143-6.1
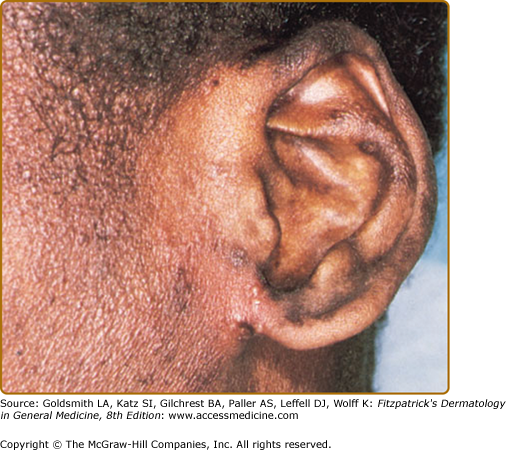
Herpetic infection with pustules on the ear of a teenager with Wiskott–Aldrich syndrome. The patient was blind in this left eye owing to previous ocular infection. After the pictured infection, the patient was administered prophylactic acyclovir and had no subsequent herpetic infections for a decade. Eventually, he became resistant to acyclovir and succumbed to his immunodeficiency.
As many as 40% of patients with WAS develop an autoimmune disorder.81 The most common are vasculitis (particularly involving the skin, GI tract, brain, and heart) in 20% of patients, autoimmune hemolytic anemia in 14%, and IgA nephropathy in up to 10% of patients.82 Other immune-mediated cutaneous manifestations are angioedema, dermatomyositis, pyoderma gangrenosum, and erythema nodosum.81 Hepatosplenomegaly is common, and lymphadenopathy, transient arthritis, and joint effusions are present occasionally.
The thrombocytopenia of WAS is persistent, and platelet counts may range from 1,000 to 80,000 platelets per μL. A platelet count of <70,000 is required for formal diagnostic criteria. The platelets are small, and platelet aggregation is defective. Levels of IgM and sometimes, IgG, are low, and isohemagglutinins are absent. IgA, IgE, and IgD levels usually are elevated. Eosinophilia, leukopenia, and lymphopenia are also seen. Delayed hypersensitivity skin-test reactivity is diminished, and patients fail to respond to polysaccharide antigens. WASp can be detected by flow cytometry using intracellular staining with an antibody to WASp and sequencing of the WASP gene can confirm the diagnosis of WAS or X-linked thrombocytopenia, which results also from mutations in the WASP gene that do not affect immune function.76 Mutation analysis allows for prenatal diagnosis.83
Therapeutic interventions allow some patients with WAS to survive into adulthood; however, a significant proportion die before the age of 10 years due to infections secondary to hemorrhage, malignancies, or the complications of transplantation.75,84,85 Thirteen percent of patients with WAS develop lymphoreticular malignancies,80 especially non-Hodgkin lymphoma, with a predominance of extranodal and brain involvement. Development of autoimmune hemolytic anemia is a poor prognostic factor and associated with the development of lymphoid malignancies; overall 25% of patients with autoimmunity develop a malignancy.82 Ten percent of patients die from these malignancies, usually as adolescents or young adults.
Appropriate antibiotics, immunizations, and transfusions of platelets and plasma decrease the risk of fatal infections and hemorrhage. Ig replacement therapy is useful in some patients. Splenectomy has been advocated to ameliorate the bleeding abnormality in patients with recurrent severe hemorrhage, but this procedure increases the risk of infection from encapsulated bacterial organisms. Bone marrow or stem cell transplantation is the treatment of choice for patients with recurrent problems, especially significant autoimmunity. Full engraftment results in normal platelet number and function, normal immunologic status, and clearance of the dermatitis (T lymphocyte engraftment). The 5-year survival rate with HLA-matched sibling donors is 87%.80 Topical glucocorticoid preparations and Ig replacement may improve the dermatitis, and chronic administration of oral acyclovir is appropriate for patients with recurrent eczema herpeticum.
|
 Severe combined immunodeficiency (SCID) includes a group of heterogeneous disorders characterized by similar clinical manifestations and immunologic deficiencies of both humoral and cell-mediated immunity.86–88 The overall incidence of SCID is 1 in 75,000 births.89 The mode of inheritance is either X-linked or autosomal recessive.
Severe combined immunodeficiency (SCID) includes a group of heterogeneous disorders characterized by similar clinical manifestations and immunologic deficiencies of both humoral and cell-mediated immunity.86–88 The overall incidence of SCID is 1 in 75,000 births.89 The mode of inheritance is either X-linked or autosomal recessive.
 All patients with SCID share most clinical features and have abnormalities of both cell-mediated and humoral immunity, although the extent of deficiency is variable. The underlying basis of SCID is the absence of T cells or the absence of T cell function. B cells and NK cells may or may not be present and SCID can be categorized based on the presence or absence of all three types of lymphocytes. The underlying genetic defect (Table 143-4) also correlates with the cellular phenotype. Ninety-five percent of SCID cases have an identified gene defect.90 SCID with absence of all three cell types usually results from reticular dysgenesis or from the accumulation of toxic metabolites in patients with mutations in the gene for the purine-degradation enzyme adenosine deaminase (ADA, 16% of patients) or purine nucleoside phosphorylase (PNP).91 X-linked SCID affects approximately 46% of patients, and is characterized by the absence of T cells and NK cells. B cells are present in normal or slightly reduced numbers. The genetic defect is a mutation in the gene encoding the γc chain of the IL-2 receptor, which is also shared with five other interleukin receptors, including the interleukin 7 receptor (IL-7R) and IL-15R, which are required for T cell and NK cell development, respectively. Janus kinase 3 is a signaling molecule downstream of the γc chain and is mutated in autosomal recessive SCID with a similar cellular phenotype as X-linked SCID. In contrast, mutations in the gene encoding interleukin 7 receptor α chain (IL-7Rα) only affects T cell development and patients with this form of SCID have no T cells but have normal numbers of B cells and NK cells.92 Genetic defects that affect the recombination of antigen receptors on T cells and B cells including gene defects in recombination-activating gene 1 or 2 (RAG1 or RAG2) or Artemis gene result in SCID with no T cells or B cells but normal numbers of NK cells.89
All patients with SCID share most clinical features and have abnormalities of both cell-mediated and humoral immunity, although the extent of deficiency is variable. The underlying basis of SCID is the absence of T cells or the absence of T cell function. B cells and NK cells may or may not be present and SCID can be categorized based on the presence or absence of all three types of lymphocytes. The underlying genetic defect (Table 143-4) also correlates with the cellular phenotype. Ninety-five percent of SCID cases have an identified gene defect.90 SCID with absence of all three cell types usually results from reticular dysgenesis or from the accumulation of toxic metabolites in patients with mutations in the gene for the purine-degradation enzyme adenosine deaminase (ADA, 16% of patients) or purine nucleoside phosphorylase (PNP).91 X-linked SCID affects approximately 46% of patients, and is characterized by the absence of T cells and NK cells. B cells are present in normal or slightly reduced numbers. The genetic defect is a mutation in the gene encoding the γc chain of the IL-2 receptor, which is also shared with five other interleukin receptors, including the interleukin 7 receptor (IL-7R) and IL-15R, which are required for T cell and NK cell development, respectively. Janus kinase 3 is a signaling molecule downstream of the γc chain and is mutated in autosomal recessive SCID with a similar cellular phenotype as X-linked SCID. In contrast, mutations in the gene encoding interleukin 7 receptor α chain (IL-7Rα) only affects T cell development and patients with this form of SCID have no T cells but have normal numbers of B cells and NK cells.92 Genetic defects that affect the recombination of antigen receptors on T cells and B cells including gene defects in recombination-activating gene 1 or 2 (RAG1 or RAG2) or Artemis gene result in SCID with no T cells or B cells but normal numbers of NK cells.89
Lymphocyte Profile | Disorder | Gene | Gene Product Function |
|---|---|---|---|
T−/B−/NK− | ADA deficiencya,b PNP deficiency | ADA PNP | Enzyme necessary for detoxification of metabolic products of the purine salvage pathway that cause lymphocytes to undergo apoptosis Enzyme necessary for detoxification of metabolic products of the purine salvage pathway that cause lymphocytes to undergo apoptosis: located downstream from ADA |
T−/B−/NK+ | RAG 1 or 2c Artemis deficiencyd,e LIG4 syndromee,93 | RAG1/RAG2 Artemis LIG4 | Required for somatic V, D, and J rearrangements during Band T-cell development Required for DNA repair during somatic V, D, and J rearrangements during B- and T-cell development Ligase required for ATP-dependent repair of double-stranded DNA |
T−/B+/NK− | X-linked severe combined immunodeficiencya,f Janus protein kinase deficiencyf | IL2RG JAK3 | Common γ chain of IL-2, -4, -7,-9,-15, and -21 Tyrosine kinase; primary signal transducer from common γ chain |
T−/B+/NK+ | IL-7 receptor (IL-7R) deficiency T-cell receptor/CD3 complex deficiency CD45 deficiency92 Winged helix nude deficiency | IL7R CD3δ and CD3ϵ PTPRC WHN | α Chain of IL-7 receptor Facilitates expression of T-cell receptor/CD3 complex; block at pre-T-cell receptor stage of development Phosphatase that regulates immune cell signaling Defect in thymic development. Human homolog of nude mouse with total alopecia and onychodystrophy; critical transcription factor for thymic development |
Other defects in T cell development | |||
CD4+ CD8−/B+/NK+ | CD8 deficiency94 ZAP-70 deficiency MHC class I deficiency/TAP deficiency (bare lymphocyte syndrome I) | CD8A ZAP-70 TAP1 TAP2 | Promotes survival and differentiation of activated T cells to memory CD8+ T cells Signaling tyrosine kinase Transporter associated with antigen processing, transports peptides to assemble the class I molecule, dysregulated NK cells |
CD4low CD8+/B+/NK+ | MHC class II deficiency (bare lymphocyte syndrome II) | CIITA RFXB RFX5 RFXAP | Defects in the transactivating factors of MHC II molecules, nonfunctional CD4 T cells if present |
T+/B+/NK+ | T-cell receptor/CD3 complex deficiency | CD3γ | Chain facilitates expression of T-cell receptor/CD3 complex |
 Mutations in the genes that encode the CD3δ and CD3ϵ chains of the T-cell antigen receptor/CD3 complex account for less than 1% of cases.89 Mutations in the gene for CD45 (phosphatase that regulates immune cell signaling),92 LIG4 (ligase required for DNA repair),93 and CD8A (which enables differentiation of memory CD8+ cells)94 also cause SCID. These molecular defects all result in total or selective lymphopenia. Additional defects fail to completely block T-cell differentiation but the resultant T cells are functionally impaired. These include ZAP-70 deficiency,95 CD3γ chain deficiency,96 MHC class II deficiency and MHC class I deficiency/transporter for antigen presentation (TAP) deficiency. The human homolog of the nude mouse with total alopecia and nail dystrophy has been linked to mutations in WHN (winged helix nude) (see Table 143-4).97
Mutations in the genes that encode the CD3δ and CD3ϵ chains of the T-cell antigen receptor/CD3 complex account for less than 1% of cases.89 Mutations in the gene for CD45 (phosphatase that regulates immune cell signaling),92 LIG4 (ligase required for DNA repair),93 and CD8A (which enables differentiation of memory CD8+ cells)94 also cause SCID. These molecular defects all result in total or selective lymphopenia. Additional defects fail to completely block T-cell differentiation but the resultant T cells are functionally impaired. These include ZAP-70 deficiency,95 CD3γ chain deficiency,96 MHC class II deficiency and MHC class I deficiency/transporter for antigen presentation (TAP) deficiency. The human homolog of the nude mouse with total alopecia and nail dystrophy has been linked to mutations in WHN (winged helix nude) (see Table 143-4).97
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


