Genetic Deposition Disorders
Titilola Sode
Adnan Mir
LIPOID PROTEINOSIS
Definition and Epidemiology
Lipoid proteinosis (LP, hyalinosis cutis et mucosae) is a rare autosomal recessive disease characterized by thickening of the skin and mucosal surfaces. Patients classically present with a hoarse voice early in childhood, variable scarring, and cutaneous yellow beaded papules. It is caused by mutations in extracellular matrix protein 1 (ECM1).
The majority of patients are of European ancestry, especially among the Dutch South Afrikaans owing to a founder effect. Fewer than 500 cases have been reported in the literature to date, with more than 50 different reported mutations in ECM1.1,2 Males and females are equally affected. Symptoms typically appear early during childhood.
Etiology
The ECM1 protein is normally found within the basement membrane zone and acts as a scaffolding molecule, binding to numerous structural proteins, including collagen IV and laminin 332.3 It also appears to have functions in epidermal differentiation and in angiogenesis. In patients with LP, loss of functional ECM1 results in hyalinized deposition of protein material at the dermoepidermal junction and within the papillary dermis.
There are several different splice isoforms of ECM1, which incorporate different exons. Some mutations cause more severe manifestations of the disease on the basis of which isoform they produce.3,4 Immune-mediated acquired dysfunction of ECM1 has been implicated in the pathogenesis of lichen sclerosus et atrophicus.
Clinical Presentation
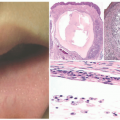
A hoarse or weak cry is the first identifiable feature of the disease, typically manifesting at birth or within the first few years, and progressively worsening throughout an individual’s life. The cutaneous features of the disease vary with age. Infants and young children typically develop vesicles or bullae with hemorrhagic crusting on the face and upper extremities and within the mouth—often secondary to trauma. These lesions heal with varioliform scarring (Figure 10-1A).
Later in childhood, there is an increase in hyaline deposition within the dermis leading to the development of yellow papules and plaques on the face, eyelids, neck, and hands. The scalp can also be affected, resulting in patchy or diffuse hair loss. Thick, waxy yellow plaques favor flexural surfaces, whereas verrucous plaques favor extensor surfaces. In about 50% of individuals, a string of bead-like papules appears on the free eyelid margins, often leading to a loss of lashes (Figure 10-1B and C). Involvement of the eyelid margin may be complicated by corneal ulcers. A cobblestone appearance may appear on mucosal surfaces within the mouth.
Systemic complications of extensive hyaline deposition include movement-limiting woody induration of the tongue, dysphagia, respiratory obstruction, premature tooth loss, parotid pain and swelling, and neurologic complications of grand mal seizures and behavioral changes.
A pathognomonic finding on radiographic examination is bilateral, intracranial, sickle or bean-shaped calcification within the temporal lobes.5
A pathognomonic finding on radiographic examination is bilateral, intracranial, sickle or bean-shaped calcification within the temporal lobes.5
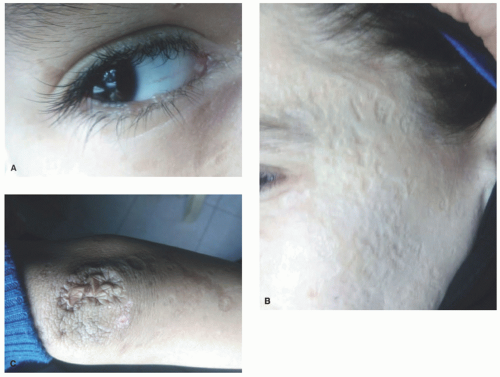 FIGURE 10-1. Lipoid proteinosis. A, Beaded, waxy papules along the eyelid margins. B and C, Characteristic, atrophic, varioliform scarring on the face and elbows. |
Histologic Findings
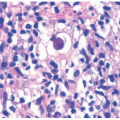
There is deposition of pale, eosinophilic, amorphous material surrounding small capillaries and eccrine ducts in the superficial dermis. In more developed lesions, deposits undergo hyaline thickening with an “onion-skin” appearance. Sweat glands are damaged with increased hyaline deposition (Figure 10-2). Hyperkeratosis and papillomatous may be seen.6 The hyaline deposits are periodic acid-Schiff (PAS) positive and diastase resistant, and stain with Alcian blue as well as Sudan black on frozen sections.7
Differential Diagnosis
The clinical differential diagnosis of LP includes xanthoma, amyloidosis, colloid milium, leprosy, and scleromyxedema.
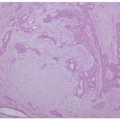
Histopathologically, there is significant overlap with erythropoietic protoporphyria (EPP) (Figure 10-3). However, these can be differentiated by the deeper, more extensive involvement of hyalinization in LP, and clinically by the distribution in more sun-exposed areas in EPP. Colloid milium and amyloidosis may be considered, but like EPP, are also typically more superficial.
CAPSULE SUMMARY
LP is a rare genetic condition resulting in hyaline deposition in the skin and mucosae. It causes diffuse waxy papules, nodules, and plaques on the head and upper extremities, as well as mucosal surfaces and in severe cases, internally. Histologically, it is characterized by the deposition of amorphous hyaline material within the dermis surrounding vessels and eccrine glands.
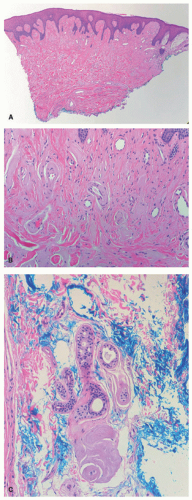 FIGURE 10-2. Lipoid proteinosis. Low- (A) and high-power (B) images showing diffuse deposits of amorphous hyaline material with a thickening of vessel walls. (C) There is involvement of the eccrine coil in this example. Courtesy of Dr. Travis Vandergriff, Department of Dermatology, UT Southwestern Medical Center. |
GAUCHER DISEASE
Definition and Epidemiology
Gaucher disease is an autosomal recessive lysosomal storage disease caused by mutation in β-glucosidase. There is resultant accumulation of the glycolipid glucocerebroside, which leads to a variety of complications affecting skin, bones, and liver.
Gaucher disease is a relatively common genetic disease that is classically divided into three clinical subtypes. Type 1, the most common, affects approximately 1/800 births in the Ashkenazi Jewish population, although it is rare in the general population with an incidence of 1 in 40 000 to 60 000.8,9 Types 2 (which has the most cutaneous findings) and 3 are rare. Males and females are affected equally.
Etiology
Gaucher disease is a sphingolipidosis, a member of the lysosomal storage group of diseases. It is caused by a mutation in β-glucosidase (glucocerebrosidase), which leads to a decrease in the enzyme’s ability to break down glucocerebroside. Its accumulation in macrophages results in the formation of Gaucher cells, which in turn accumulate within bone and lead to bone destruction. The pathophysiology of Gaucher disease in the skin is poorly understood, but is likely related to lipid processing and cell membrane formation, much like many of the congenital ichthyoses. Ultrastructural studies of skin from patients with Type 2 Gaucher disease support this explanation.10
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


