Achondroplasia
Hypochondroplasia
Pseudoachondroplasia
Description
Rhizomelic shortening secondary to short humeri, flexion contractures from flexion deformities of distal humerus, elbow abnormalities, and trident appearance of hand [extra space between third and fourth rays]. Short stature noticeable at birth, foramen magnum stenosis, thoracolumbar kyphosis, spinal stenosis, and genu varum [14, 17]
Defective conversion of cartilage to bone. Less severe form of achondroplasia—body changes milder and often overlooked. Normal trunk length, disproportionately short arms and legs, hands and feet are broad and short. Differentiated from achondroplasia by lack of facial dysmorphism, less severe short stature, less obvious skeletal disproportion, and milder radiologic findings [17, 20]
Moderate to severe disproportionate short stature, ligamentous laxity, and progressive degenerative joint disease. Short-limb dwarfism with epiphyseal and metaphyseal involvement. Moderate brachydactyly, joint hyperextensibility in hands, restricted extension at elbows, and overall joint pain. Osteoarthritis in early adulthood [21–23]
Radiographic findings: Same as achondroplasia, but milder [18]
Genetics
Autosomal dominant; Locus—4p16.3; Gene—FGFR3; Protein—FGFR3 [24]
Autosomal dominant; Locus—19p12-13.1; Gene—COMP; Protein—Cartilage Oligomeric Matrix Protein (COMP) [24]
Natural History
Normal length and facies at birth. Often presents at the onset of walking with a waddling gait. By 2 years of age, growth rate below the standard growth curve which leads to disproportionate short-limb short stature. Average adult heights: 116 cm for females and 120 cm for males [21]
Treatment
Growth hormone therapy and limb lengthening if necessary [26]
Evaluate for skeletal manifestations. Anterior/posterior radiographs of hands. Assess ligamentous laxity. Analgesics for joint pain [21]
Thanatophoric dwarfism/dysplasia type 1
Thanatophoric dwarfism/dysplasia type 2
Marfan syndrome
Description
Genetics
Autosomal dominant; Locus—4p16.3; Gene—FGFR3; Protein—FGFR3 [24]
Autosomal dominant; Locus—15q21.1; Gene—FBN1; Protein—Fibrillin-1 [24]
Natural History
Most common type of lethal neonatal skeletal dysplasia; overall association with rhizomelic limb shortening, macrocephaly, and cloverleaf skull. Difficult to differentiate from other forms of short-limb dwarfism—most important difference is that TD has severe rib shortening, restricted lung volume, and respiratory distress leading to death within a few hours of birth. Most infants do not survive past a few hours or days due to respiratory insufficiency [27–29]
Children taller than average for age. By adulthood, may reach 7 ft tall [19]
Treatment
At birth, infant may require suboccipital decompression to alleviate craniocervical junction constriction. Joint contractures or joint hypermobility should be evaluated and followed [29]
Therapy with nighttime splinting in extension is often successful for treatment of camptodactyly. More severe cases may require tendon transfer, release of volar structures and PIPJ. Surgery outcomes are unpredictable [30]
Osteogenesis imperfecta
Nail–Patella syndrome
Diastrophic dysplasia
Description
Endochondral ossification affected causing short stature from shortened limbs, progressive spinal deformities, foot deformities, frequent joint subluxation and dislocation, large joint contractures, ear pinnae deformities, and/or cleft palate. Hitchhiker thumb, shortened fingers, synostosis of the proximal interphalangeal joints, and ulnar deviation of fingers. Radial dislocation may also be seen clinically [33–35]
Radiographic findings: Osteopenia, bone fractures, and bone deformities [31]
Radiographic findings: Radial head and capitellum hypoplasia, elbow dislocation [18]
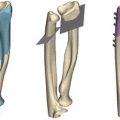
Radiographic findings: Micromelia; short, thick tubular bones; epiphyseal dysplasia; metaphyseal flaring of long bones; bifid or V-shaped distal humerus, may also be pointed and hypoplastic; radial bowing; proximal radial dislocation at birth; brachydactyly and short ovoid first metacarpal; irregular carpal bones; joint dislocations [18, 33]
Genetics
OI Types I–IV: Autosomal dominant; Gene—COL1A1 or COL1A + G42; Protein—type 1 collagen
Autosomal dominant; Locus—9q34.1; Gene—LMX1B; Protein—LIM homeobox transcription factor 1 [24]
Autosomal recessive; Locus—5q32-33; Gene—DTDST; Protein—SLC26A2 sulfate transporter [24]
OI Type V: Autosomal dominant; Gene—unknown
OI Type VI: Autosomal recessive; Gene—unknown
OI Type VII: Autosomal recessive; Protein—Cartilage-associated protein [CRTAP] [24]
Natural History
More severe forms of OI may experience bone fragility and fracture in utero and/or at birth. Milder forms may remain nearly absent in adulthood. Overall, fracture incidence decreases after puberty and increases after menopause and males in their 60s [1]
Diagnosis can be made through ultrasound and molecular genetic testing prenatally or clinically at birth. Normal mental status. Growth and motor capabilities greatly affected by deformities. Disproportionate dwarfism with a mean height of 130–140 cm can be seen in affected adults [36]
Treatment
Bisphosphonates may be used to decrease fracture frequency, improve vertebral bone density, and strengthen grip. Surgical goal is to minimize fracture frequency, restore bone axis, and avoid bowing. Long bone internal fixation in children is common via multilevel osteotomies and telescopic intramedullary nail fixation. Long-term rod revision surgery may be required [3, 4]
Focus on improving mobility through casting to maintain joint positioning, physiotherapy, and other forms of therapy. Cervical spinal surgery only indicated with clinical or neurophysiological evidence of spinal cord impingement—otherwise, cervical kyphosis typically spontaneously corrects. In cases of premature degenerative arthrosis, arthroplasty is indicated. Early physical therapy may prevent joint contractures [33, 35]
Kniest’s dysplasia
Cleidocranial dysostosis/dysplasia
Niemann–Pick disease
Description
Lipid storage disease. Previously not known to have skeletal involvement. Joint and/or limb pain has been reported as well as decreased bone mineral density [BMD] in both affected pediatric and adult patients [39]
Radiographic findings: Multiple pseudoepiphyses of metacarpals and tapered distal phalanges in hands [18]
Genetics
Autosomal dominant; Locus—12q13.1; Gene—COL2A1; Protein—type 2 collagen [24]
Autosomal dominant; Locus—6p21; Gene—RUNX2; Protein—Run related transcription factor 2 [24]
Natural History
Mean adult height for males is 162 cm [26]
Patients with neurological involvement do not survive beyond 3 years. Patients without neurodegeneration usually survive into late childhood or adulthood [39]
Treatment
UE management not well documented
Orthopedic intervention may be necessary if severe impairment or disability occurs [38]
No definitive treatment. Early intervention for low BMD such as load-bearing activities and muscle strengthening exercises. Frequent pulmonary disease and chronic fatigue must be considered [39]
Mucopolysaccharidoses
Description
Defective endochondral and membranous growth. Presents with dysostosis multiplex—short stature, platyspondyly with anterior beaking, ‘bullet-shaped’ phalanges. Joint contractures and carpal tunnel syndrome are common. Osteopenia may occur in association with pathologic fractures
MPS I H—Hurler syndrome: Carpal tunnel syndrome, joint contractures, and dysostosis multiplex
MPS I S—Scheie syndrome: Carpal tunnel syndrome, joint contractures, and dysostosis multiplex
MPS II—Hunter syndrome: only X-linked MPS disorder, Carpal tunnel syndrome, joint contractures, and dysostosis multiplex
MPS IIIA-B—Sanfilippo Types A-B: Less severe than I, II, VI, and VII
MPS IVA—Morquio Type A: severe skeletal dysplasia, joint hypermobility, and dysplastic odontoid process
MPS IVB—Morquio Type B: severe skeletal dysplasia, joint hypermobility, and dysplastic odontoid process
MPS VI—Maroteaux–Lamy syndrome: Carpal tunnel syndrome, joint contractures, and dysostosis multiplex
Radiographic findings: Coarsened long bones, shortened ulna, Madelung deformity of distal radius, shortened metacarpals with proximal tapering, and broad clavicles [42]
Genetics
Autosomal recessive; Gene—varies by type of MPS [42]
Natural History
Treatment
Hereditary multiple exostoses/multiple osteocartilaginous exostoses/diaphyseal aclasia
Fibrodysplasia ossificans progressiva
Chondroectodermal dysplasia/Ellis–van Creveld syndrome
Description
Multiple cartilage-capped boney protuberances, or osteochondromas, at metaphyses of long bones. Mild short stature and disproportionate short-limbs. Rarely, an enchondroma may undergo a malignant transformation into secondary chondrosarcoma. UE most commonly presents with length discrepancy between the radius and ulna—radial bowing, radial tilting, and radial head dislocation may occur [43]
Fibrous tissues, muscles, and periosteal regions undergo progressive ossification. Shortened and deformed thumbs [19]
Short stature, irregular bone growth ad structure. Polydactyly also occurs [19]
Genetics
HME–1: Autosomal dominant, Locus—8q23-24.1; Gene—EXT1; Protein—Exostosin-1
Autosomal dominant; Locus—4q27-31, 17q21-22, or 2q23-24 [19]
Autosomal recessive; Locus—4p16 [19]
HME–2: Autosomal dominant, Locus—11p12-11; Gene—EXT2; Protein—Exostosin-2
Natural History
Numerous osteochondromas develop near growth plates. During childhood and adolescence, osteochondromas create a pseudo-growth plate and cause deformity with growth [44]
At age five, patient starts developing large ectopic osseous collections in muscular regions. These osseous collections cause severe disability and limits joint movement [19]
Treatment
Growth deformity correction and removal of symptomatic osteochondromas. To manage impending or complete radial head dislocation: Ulnar collateral carpal ligament release at the wrist and radial head resection at skeletal maturity. Ulnar wrist deviations are usually asymptomatic. If not, acute and guided-growth interventions may be successful. Malignant transformation into chondrosarcoma must be resected. Typically low grade [43]
No known effective treatment. Surgery, corticosteroids, and radiotherapy have been used. Bisphosphonates have been used to decrease ectopic osseous masses but clinical benefits are not well established [45]
Surgical excision of polydactyly [30]
Ehlers Danlos syndrome (EDS)
Spondyloepiphyseal DYSPLASIA
Multiple epiphyseal dysplasia
Description
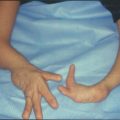
Connective tissue disorder characterized by congenital joint hypermobility, skin hyperextensibility, and tissue fragility. Joint dislocations due to little to no trauma are common as is chronic limb pain. Severity varies with type of EDS [46, 47]
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree

 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access
