Fabry Disease: Introduction
|
Epidemiology
Fabry disease (α-galactosidase A, GLA, deficiency) is generally considered the second most prevalent lysosomal storage disorder, after Gaucher disease1,2 with an estimated incidence ranging between 1:40,000 to 1:170,000 persons. All ethnic groups are affected. Milder variants of the disease are associated with significant residual enzyme activity and may occur with much higher frequency (e.g., 1:3,200), as suggested by a recent survey of neonates in Northern Italy.3 These variants often have predominant involvement of a single organ (e.g., heart or kidney) and have onset in adult life.
Etiology and Pathogenesis
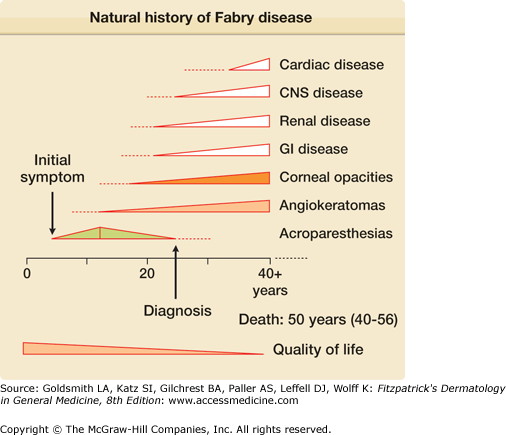
Fabry disease (OMIM #301500) is caused by the partial or complete deficiency of a lysosomal enzyme, α-galactosidase A (Fig. 136-1). As a result of this enzyme deficiency, neutral sphingolipids with terminal α-galactosyl residues [predominantly globotriaosylceramide (Gb3)] accumulate in the lysosomes of different tissues and fluids (epithelial cells of glomeruli and tubules of the kidneys; cardiac myocytes; ganglion cells of the autonomic system; cornea; endothelial, perithelial, and smooth muscle cells of blood vessels; and histiocytic and reticular cells of connective tissue). Clinical onset is variable (Fig. 136-2). Although the condition is X linked, heterozygous females are frequently affected and may be as severely affected as hemizygous males.4 Clinical symptoms typically occur a decade or so later in females than in males and organ damage is usually less severe. Variable expression in females is attributed to the Lyon hypothesis, whereby one X chromosome is inactivated on a random basis in the female, while the other provides the genetic information.
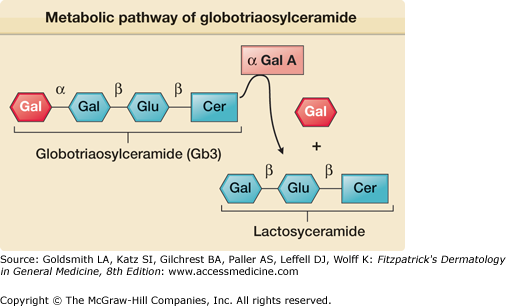
The GLA gene for α-galactosidase A is located at the Xq22.1 region. More than 400 mutations have been identified in the GLA gene,5,6 including missense, nonsense mutations, and single amino acid deletions and insertions. Most of these mutations are “private,” having been identified only in individual families. Some (e.g., the p.N215S mutation) have been associated with single organ or late onset variants and patients have normal levels of urinary Gb3, despite having clinical evidence of Fabry disease.
Historical Aspects
Cutaneous Manifestations
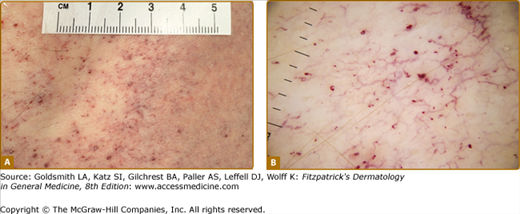
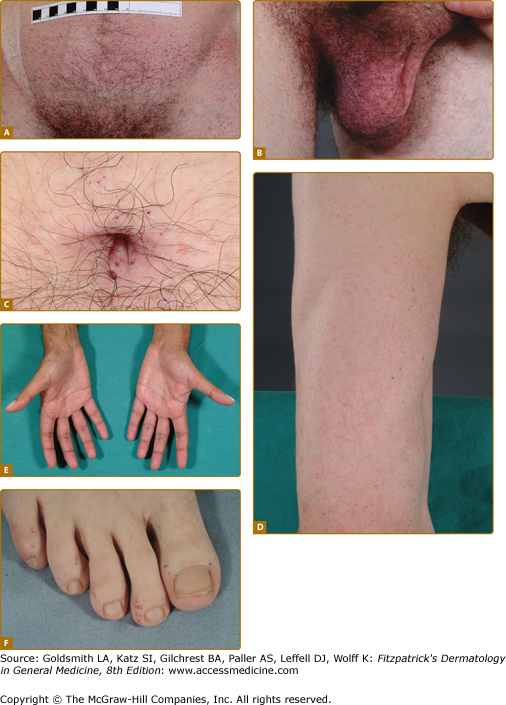
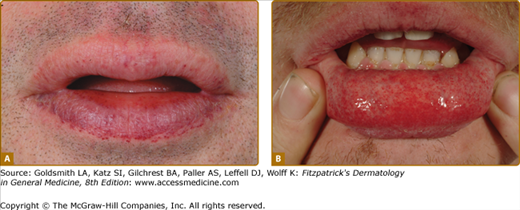
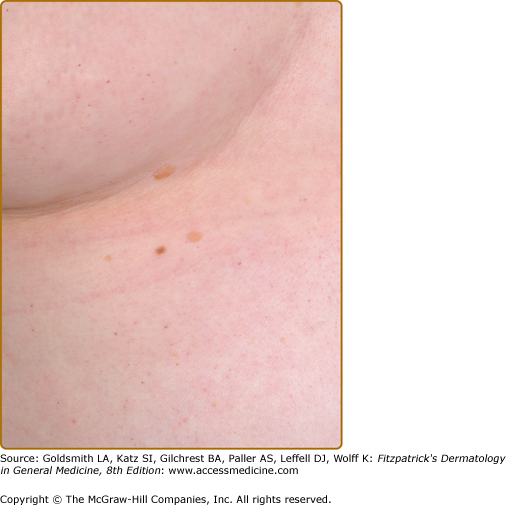
Angiokeratoma (AK), the cutaneous hallmark of Fabry disease,7,8 are present in 70% of males and 39% of females.9,10 They are pinpoint to 4-mm diameter, dark-red to blue–black, macular, and papular lesions, which do not blanch on pressure (Fig. 136-3). Overlying hyperkeratosis is variable and frequently absent at sites other than the genitalia and umbilicus.9,11 AKs appear between the ages of 5 and 15 years in males and 3–10 years later in females.12–15 They were a presenting feature in 14.2% of 352 pediatric patients,15 31.7% of adult males (n = 359), and 11.2% of adult females (n = 118).4 They may be widespread or grouped. In males, lesions are typically within the “bathing trunk” area (genitals, buttocks, lower abdomen, umbilicus, groins, inner thighs, and sacrum). AK are also often seen on the proximal limbs, particularly their medial aspects, the elbows and knees, the palms and soles, and over the distal phalanges of the digits (Fig. 136-4). Lesions may occur on the lips, particularly along the vermilion border, and occasionally on the mucosal surfaces (Fig. 136-5). They rarely occur elsewhere on the face. In females, AK are usually sparsely distributed (Fig. 136-6) and may occasionally occur in a dermatomal distribution.9,16 The commonest sites are the trunk and proximal limbs.9 Female genital lesions are relatively infrequent.
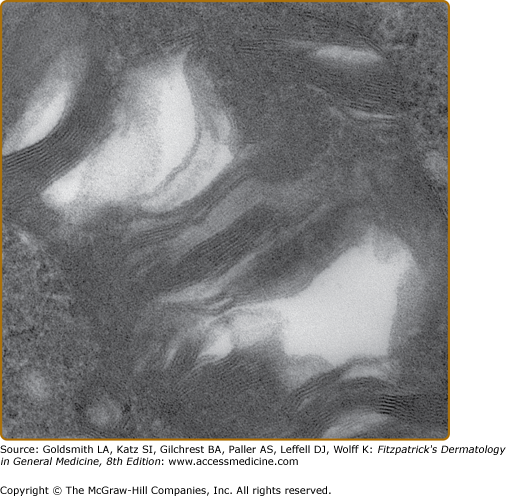
Histologic evaluation of lesional skin typically shows dilated, ectatic capillaries in the papillary dermis, a variably thinned epidermis centrally, with epidermal acanthosis at the edges of the lesion, and variable degrees of overlying focal compact orthohyperkeratosis (eFig. 136-6.1A). Endothelial, perithelial, perineural, eccrine, smooth muscle cells, and fibroblasts are filled with cytoplasmic vacuoles containing glycosphingolipid that can be visualized with toluidine blue stains. Characteristic, electron dense, lamellated intracytoplasmic vacuolar inclusions (zebra bodies) are typically seen on electron microscopy17 (eFig. 136-6.1B; Fig. 136-7; eFig. 136-7.1; and Figs. 136-8A and 136-8B). They exhibit a pattern of alternating light and dark 4–6-nm bands. These inclusions may be present in biopsies from AK or from normal skin. They may be absent in the skin of heterozygous females.17 Electron microscopy and immunoelectron microscopy using anti-Gb3 antibodies are useful tools when other diagnostic tests are not conclusive.18
eFigure 136-6.1
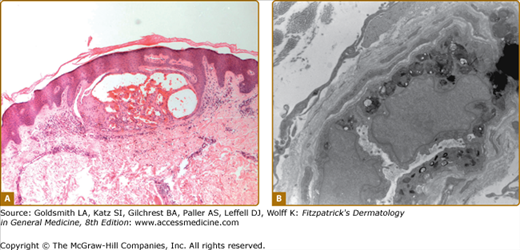
Histopathologic features of angiokeratoma. A. Light microscopy: dilated capillaries in the papillary dermis with overlying epidermal thinning and mild orthohyperkeratosis. Acanthotic epidermis at the periphery. H&E stain ×40 magnification. B. Electron microscopy: multiple electron dense inclusions in a cutaneous vascular endothelial cell magnification ×9,000.
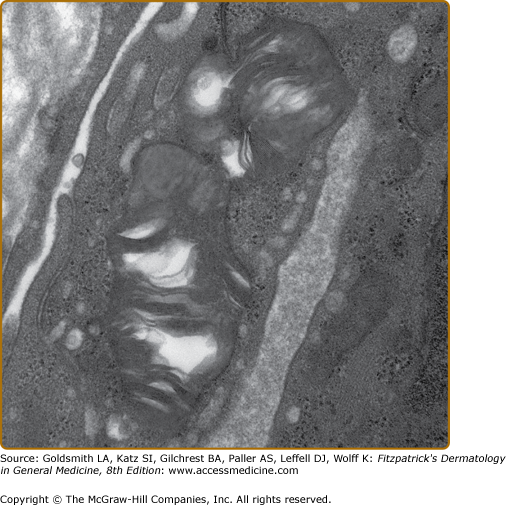
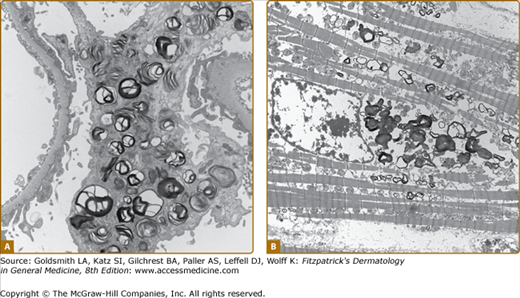
Up to a third of Fabry males and two-thirds of females do not have AKs.9,10,13,14 A proportion have no cutaneous vascular lesions (CVL),9 and others have bright red macular angiomatous lesions, which could represent early AKs or macular angiomas clinically.11 Patients with widespread papular angiomas clinically and histologically in keeping with cherry angiomas are also recognized. In one series, patients with a predominantly cardiac phenotype (N215S mutation) rarely had classical AK, but a third of males in this group had widespread (>100) cherry angiomas.19 Since the prevalence of such lesions in the general population may be as high as 50%, their significance in patients with Fabry disease is not clear. Telangiectasias are also common,9 and may be distinguished from AKs and macular and cherry angiomas by the presence of blanching on diascopic pressure. Registry data show that they are more common in males (23%) than in females (9%).9,10 Although based on patient recall, data suggest that they appear later than AK, with a mean age at onset of 26 years in males (range 3–70) and 42 years in females (range 5–73).9 In a majority of cases, telangiectasias occur at sun-exposed sites, such as the face and V of the neck, often in patients with skin type I or II. Occasionally, telangiectasias may be seen at unusual sites such as the flanks, groins, elbow, and knee flexures. Dermoscopy of these areas reveals dilated, tortuous upper dermal vessels (Fig. 136-3 and eFig. 136-8.1). This tends to occur in patients with widespread AK and full phenotypic expression of disease.
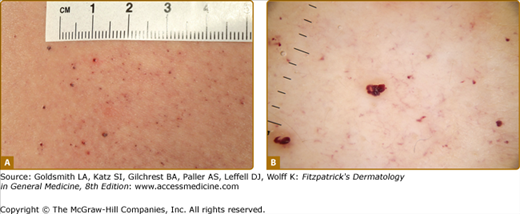
In both sexes, the presence of CVL, namely telangiectasias and/or AK, has been associated with higher disease severity scores and a higher prevalence of major organ involvement.9,10 Thus, cerebrovascular involvement (stroke and transient ischemic attacks) was present in 38% females and 32% males with CVL, but only in 12% females and 9% males without CVL. Cardiac involvement occurred in 80% females and 73% males with CVL versus 38% females and 49% males without; renal involvement in 62% females and 72% males with CVL versus 29% females and 42% males without. Similar differences are observed for hypertension, eye, ear, gastrointestinal (GI), and other neurological involvement.10 These findings demonstrate the importance of dermatological assessment and its possible predictive value in terms of systemic morbidity.
A “pseudoacromegalic” facial appearance has been described in some families with Fabry disease (Fig. 136-9 and eFig. 136-9.1). This feature is documented in two larger studies. An assessment of 38 patients by a panel of three clinical geneticists, based on standardized medical photography20 identified (in order of decreasing frequency): periorbital fullness, prominent ear lobes, bushy eyebrows, recessed forehead, pronounced nasal angle, generous nose/bulbous nasal tip, prominent supraorbital ridges, shallow midface, full lips, prominent nasal bridge, broad alar base, coarse features, posteriorly rotated ears, and prognathism. A second study, in which facial dysmorphism was objectively assessed by three-dimensional (3D) dense surface modeling and anthropometric analysis using a 3D photogrammetric camera, compared facial features in 20 males and 22 females with Fabry disease with controls.21 This study confirmed, in males and less prominently in females, the presence of the facial features noted above. The presence of these facial features should prompt appropriate investigations for Fabry disease.

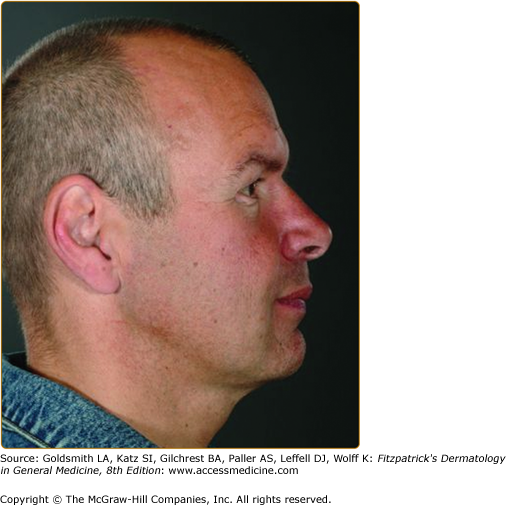
The Fabry face appears commoner in patients with widespread AKs and classical disease at the more severe end of the spectrum. Moderate or marked facial changes were present in 63% of 41 males with a classical disease phenotype, 61% of whom had AK corporis diffusum. In contrast, facial features were present to a very mild degree (periorbital puffiness only) in only 10.5% of 19 males with a predominantly cardiac phenotype of disease.10
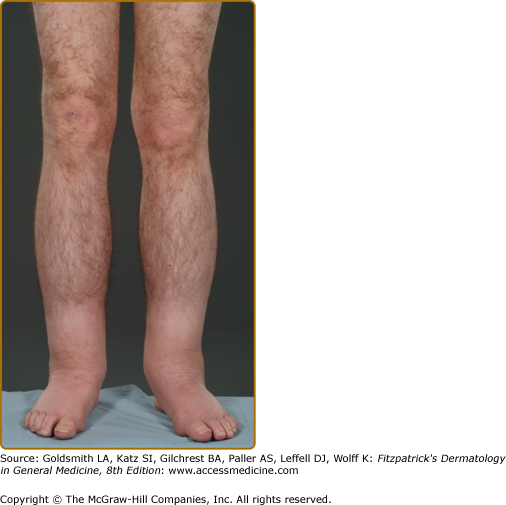
Edema and lymphedema, particularly affecting the lower limbs, is frequently observed in Fabry patients (Fig. 136-10). Lymphedema was cited in the original description of Fabry Disease (FD) and is recognized in other lysosomal storage disorders (LSDs) such as α-N-acetylgalactosaminidase deficiency.22 Case reports document it as an unusual presenting feature of the condition23 and some suggest that familial lymphedema and Fabry disease might be linked.24 Registry data from the Fabry Outcome Survey (FOS) on more than 700 patients confirm that reversible peripheral edema of the lower limbs was present in 25% of Fabry males and 17% of females; lymphedema was present in 16% of males and 7% of females. The mean age of onset was 37 years for males (range 13–70) and a decade or so later for females.9 This is significantly more than the documented UK community prevalence of 1.33/1,000.25 In the community it is five times commoner in women. The mechanism underlying the changes is unclear; their presence does not correlate with renal or cardiac involvement. Contributory factors may include glycosphingolipid accumulation,26 recurrent edema, and primary abnormality of the lymphatics.27,28 Using the technique of fluorescence microlymphography and measurement of lymph capillary pressure, Amann-Vesti et al demonstrated fragmentation of the microlymphatic network in 5/5 hemizygous males and 5/5 heterozygous females but in none of 12 healthy controls.28 Severe structural and functional changes in the initial lymphatics of the skin were present, even when lymphedema was not manifest.28 The possibility that abnormalities in expression or function of recently identified mediators of lymphangiogenesis (VEGF-C/-D and VEGF-Rs)29 may be involved in the development of lymphedema in Fabry disease remains to be investigated.
Reduced sweating is a classical feature of Fabry disease and thought largely to be a consequence of autonomic neuropathy, though substrate accumulation within sweat glands may play a role.30,31 Hypohidrosis was reported by 53% of males and 28% of females, with earlier onset in males (23 years vs. 26 years). Anhidrosis was described by 25% of males, but only by 4% of females.9 Heat intolerance is a commonly associated and disabling symptom, resulting in reduced exercise tolerance, nausea, dyspnea, light-headedness and headache, or complete collapse with loss of consciousness. Previous studies31,32 have demonstrated an improvement in sweating in Fabry patients undergoing enzyme replacement therapy (ERT). Hyperhidrosis may also occur, more commonly in females (11.9%) than in males (6.4%).33 This is higher than the estimated prevalence of 1%–2.8% of the general population in the United States.34 In a majority of Fabry patients, it affects palms and soles and is not generalized.33
“Cold intolerance” and the development of pain in the extremities in cold environments is a frequent complaint amongst Fabry patients. More recently, Raynaud phenomenon has been documented in 8% of 710 females and 11% of 644 males (data from FOS). This reversal of the sex ratio and high prevalence in males suggest a possible causal link to the underlying Fabry disease. A recent fluoroscopic nail fold capillaroscopy study35 examined 25 Fabry patients (17 males) and showed significantly more bushy capillaries and clusters in cases (72%) than controls (10%). Morphological and functional abnormalities of nail fold capillaries were present. This abnormal vasoreactivity of digital vessels could be related to autonomic dysfunction. Abnormalities of nitric oxide synthetase and increased oxidative stress in vascular endothelium and smooth muscle might also be important triggers.36
Noncutaneous Findings
Typical Time at Onset | Signs and Symptoms |
|---|---|
Childhood and adolescence (≤16 years) |
|
Early adulthood (17–30 years) |
|
Later adulthood (age >30 years) |
|








