Abstract
Until recently, the nosology of severe acute bullous disorders due to infectious agents or drugs has been confusing. It has now become clear that Stevens–Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are variants within a continuous spectrum of adverse drug reactions, whereas erythema multiforme (EM) is a distinct disorder with different clinical signs and precipitating factors, e.g. herpes simplex virus (HSV) infections. Therefore, EM is discussed separately from SJS and TEN and distinguishing features are outlined.
Keywords
erythema multiforme, Stevens–Johnson syndrome, toxic epidermal necrolysis, adverse cutaneous drug reaction, erythema multiforme minor, erythema multiforme major, SCORTEN
Erythema Multiforme
▪ Erythema multiforme minor – erythema multiforme von Hebra
- ▪
A self-limited but potentially recurrent disease
- ▪
Abrupt onset of papular “target” lesions, with the vast majority of lesions appearing within 24 hours
- ▪
Two types of target lesions are recognized: (1) typical, with at least three different zones; and (2) atypical papular, with only two different zones and/or a poorly defined border
- ▪
Target lesions favor acrofacial sites
- ▪
Erythema multiforme minor: typical and/or occasionally atypical papular target lesions with little or no mucosal involvement and no systemic symptoms
- ▪
Erythema multiforme major: typical and/or occasionally atypical papular target lesions with severe mucosal involvement and systemic features
- ▪
A preceding HSV infection is the most common precipitating factor; occasionally, there are other preceding infections or, rarely, drug exposure
- ▪
Diagnosis of erythema multiforme requires clinicopathologic correlation and is not based solely on histologic findings
- ▪
Erythema multiforme does not carry the risk of progressing to toxic epidermal necrolysis
Introduction
Erythema multiforme (EM) is an acute, self-limited skin disease characterized by the abrupt onset of symmetric fixed red papules, some of which evolve into typical and/or occasionally “atypical” papular target lesions . The eruption is often precipitated by an infection, particularly HSV. Two forms of EM are recognized – EM minor and EM major. Both are characterized by the same type of elementary lesions (targets), but are distinguished by the presence or absence of mucosal involvement and systemic symptoms ( Table 20.1 ). In the vast majority of patients, EM can be distinguished clinically from Stevens–Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) based upon the type of elementary skin lesions and their distribution .
| COMPARISON OF ERYTHEMA MULTIFORME (EM) MINOR, EM MAJOR, STEVENS–JOHNSON SYNDROME (SJS), SJS/TOXIC EPIDERMAL NECROLYSIS (TEN) OVERLAP, AND TEN | ||||||
|---|---|---|---|---|---|---|
| Type of skin lesions | Distribution | Mucosal involvement | Systemic symptoms | Progression to TEN | Precipitating factors | |
| EM minor ( Figs 20.1 & 20.2 ) |
| Extremities (especially elbows, knees, wrists, hands), face | Absent or mild | Absent | No |
|
| EM major ( Fig. 20.3 ) |
| Extremities, face | Severe | Usually present
| No |
|
| SJS ( Figs 20.7 & 20.14 ) |
| Isolated lesions Confluence (+) Trunk, face | Severe | Usually present
| Possible |
|
| SJS/TEN overlap ( Fig. 20.11 ) |
| Isolated lesions Confluence (++) Trunk, face, neck | Severe |
| Likely |
|
| TEN ( Figs 20.8 & 20.9 ) |
| Confluence (+++) Isolated lesions rare Trunk, face, neck, elsewhere | Severe, with involvement of respiratory and gastrointestinal mucosa |
|
| |
History
EM was first described by the Austrian dermatologist Ferdinand von Hebra in 1860 . The disease he described was mild with a sudden onset of hundreds of red papules. By daily observation, von Hebra recognized that some of the original papules evolved into lesions with concentric zones of color change, which he termed “target” lesions. Some of the target lesions resembled a “herpes iris”. However, von Hebra described neither a prodrome nor the presence of mucosal lesions. He recognized that the condition could be recurrent and mentioned a “typus annuus” which recurred every spring.
In 1950, Bernard Thomas further subdivided EM into EM minor and EM major . Thomas considered von Hebra’s disease to be EM minor, and severe mucosal necrosis with “EM-like” cutaneous lesions to be EM major. Since 1950, much confusion has developed regarding the definition of EM, in part because some authors, especially in the US, have stated that EM major includes SJS. However, recent investigations have clarified the situation by providing strong evidence that the term “EM minor” should be reserved for von Hebra’s disease and the term “EM major” should be used for EM associated with mucosal lesions and systemic symptoms, but not for describing SJS, because SJS and EM major are distinct clinical disorders . That said, because these are biologic processes, there can be patients where the distinction proves challenging.
Epidemiology
EM is predominantly observed in young adults and is very uncommon during childhood . There is a slight male preponderance, but no racial predilection. The exact incidence rate of EM is not known.
Pathogenesis
Current understanding suggests that EM is most likely a mucocutaneous immune reaction that occurs in the setting of an infection in certain “predisposed” individuals. HSV is clearly the most common associated infectious agent, with Mycoplasma pneumoniae (which also may be associated with SJS), Histoplasma capsulatum , and parapoxvirus (orf) observed less frequently ( Table 20.2 ) . Histoplasmosis-associated EM may occur more often in patients with concomitant erythema nodosum. To date, evidence for EBV as a precipitating factor is incomplete . Rarely, EM has been associated with drugs or systemic disease (see Table 20.2 ). Consequently, the possibility of SJS, generalized fixed drug eruption, polymorphous exanthematous drug eruption, or urticaria should be strongly considered if the presumed diagnosis is drug-induced EM . Of note, several physical agents such as trauma, cold, and UV and orthovoltage irradiation have been described as triggers for outbreaks of EM related to infectious agents, drugs or systemic disease.
| PRECIPITATING FACTORS IN ERYTHEMA MULTIFORME | ||
|---|---|---|
| Infections (approx. 90% of cases) | Viral |
|
| Bacterial |
| |
| Fungal |
| |
| Drugs (unusual) | Primarily:
| |
| Exposures (unusual) |
| |
| Systemic disease (rare) | ||
* Also precipitating factor for Stevens–Johnson syndrome and isolated oral mucositis.
† May be a pattern of cutaneous lesions in the disease rather than a precipitating factor.
To date, there is no clearly defined genetic predisposition for the development of EM. Several small studies of HLA alleles have found different associations: HLA-DQw3 (specifically the DQB1*301 split), DRw53, and Aw33 . These HLA associations differ from those reported for SJS and TEN (see below).
Herpes simplex virus and Mycoplasma pneumoniae
In the majority of children and adults with EM, the disease is precipitated by HSV types 1 and 2 . Preceding herpes labialis is noted in ~50% of patients with EM . Herpes labialis may precede the onset of the cutaneous lesions, occur simultaneously, or be evident after the target lesions of EM have appeared. Most commonly, herpes labialis precedes target lesions of EM by 3–14 days. It is presumed that most cases in children and young adults are due to HSV type 1, but documented cases of HSV type 2 in adolescents and young adults have been reported .
Most of our current knowledge regarding EM is related to the study of HSV-associated EM. Not only are HSV-encoded proteins found within affected epidermis , but HSV DNA can be detected within the early red papules or the outer zone of target lesions in 80% of individuals with EM . The presence of fragments of HSV DNA (most often comprised of sequences that encode its DNA polymerase) within the cutaneous lesions, as well as the expression of virally encoded antigens on keratinocytes, may be interpreted as evidence for replicating HSV within affected skin sites . However, replication must be at a low level, because usually HSV cannot be cultured from EM lesions.
The inflammation within cutaneous lesions is believed to be a part of an HSV-specific host response . Individuals with HSV-associated EM have normal immunity to HSV, but may have difficulty clearing the virus from infected cells; within sites of cutaneous lesions, HSV DNA may persist for 3 months after a lesion has healed . Development of cutaneous lesions is initiated by the expression of HSV DNA sequences within the skin, followed by the recruitment of virus-specific T helper type 1 (Th1) cells that produce interferon-γ in response to viral antigens within the skin . An “autoimmune” response is then thought to result from the recruitment of T cells that respond to autoantigens released by lysed/apoptotic viral antigen-containing cells. More recently it was shown that the HSV DNA fragments described above are transported (by peripheral blood CD34 + Langerhans cell precursors) to the sites where EM skin lesions will develop prior to each eruption .
While HSV is the predominant cause of EM, there are other associated infectious agents. In particular, a severe acro-mucosal presentation with mucositis, conjunctivitis, and targetoid or bullous skin eruptions can be seen in patients with M . pneumoniae infections, primarily community-acquired pneumonia. This variant occurs most commonly in young boys and adolescents. M. pneumoniae has been cultured from the bullae in patients with EM-like skin reactions, suggesting an etiologic role . However, this association could also be explained by autoimmune molecular mimicry, as in M. pneumoniae -related Guillain–Barré syndrome .
Clinical Features
Due to a certain degree of clinical similarity, EM minor, EM major, SJS and TEN have, until recently, all been considered to be part of a single disease spectrum. However, as discussed previously, there is now strong evidence to support the concept that EM is a disease distinct from SJS and TEN at several levels – clinical, prognostic, and etiologic. Clinical criteria allow the distinction of both forms of EM from SJS/TEN in the vast majority of patients . These clinical criteria are as follows: (1) the type of elementary skin lesion; (2) the distribution of skin lesions (topography); (3) the presence or absence of overt mucosal lesions; and (4) the presence or absence of systemic symptoms (see Table 20.1 ).
Elementary skin lesions ( Fig. 20.1 )
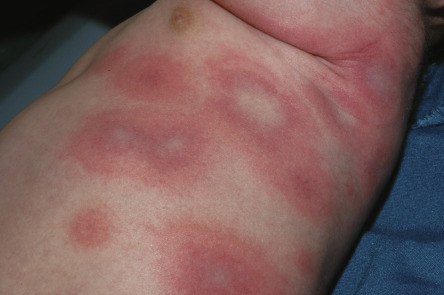
The characteristic elementary skin lesion of EM is the typical target lesion . The latter measures <3 cm in diameter, has a regular round shape and a well-defined border, and consists of at least three distinct zones, e.g. two concentric rings of color change surrounding a central circular zone that has evidence of damage to the epidermis in the form of bulla formation or crust . Frequently, this central circular zone has a dusky appearance and over time, the lesion may resemble a “bull’s eye”.
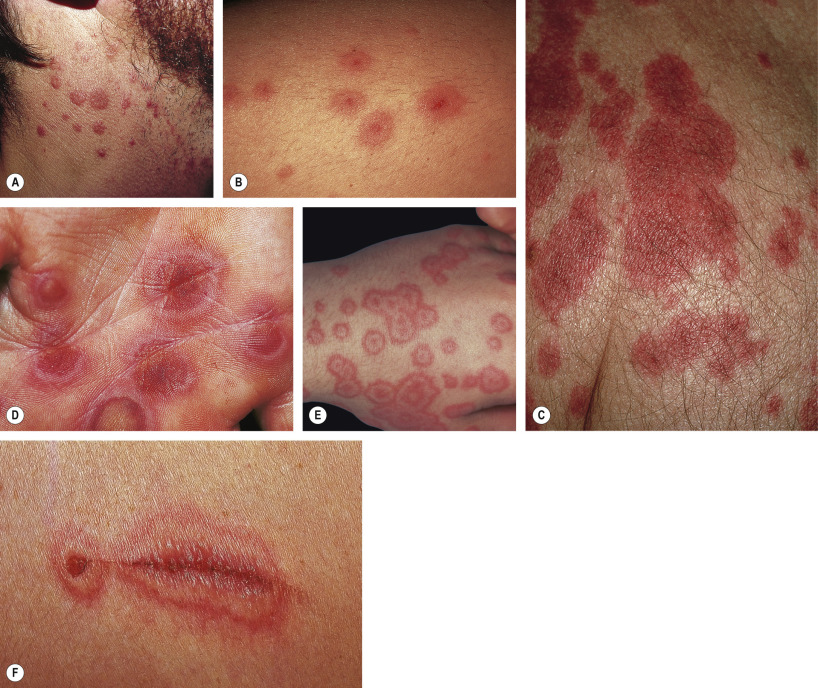
While early target lesions often have a central dusky zone and a red outer zone (“iris” lesion), they can evolve to three zones of color change. Each concentric ring within the target lesion most likely represents one of a sequence of events of the same ongoing pathologic process. This may explain why some patients have only a limited number of fully developed, typical targets amidst a number of target lesions that are not yet typical or fully evolved, while in others all the lesions are at the same stage of development, thus creating a monomorphic clinical appearance. Given the possibility that only a few typical target lesions may be present, a complete skin examination is essential.
In EM, elevated atypical papular target lesions can either accompany typical target lesions or constitute the primary cutaneous lesion. These particular lesions are defined as round, edematous, palpable and reminiscent of EM, but with only two zones and/or a poorly defined border. They must be distinguished from the flat (macular) atypical targets that are seen in SJS or TEN, but not EM. The latter are defined as round lesions that are also reminiscent of EM, but with only two zones and/or a poorly defined border, as well as being non-palpable (with the exception of a potential central vesicle or bulla).
Distribution of skin lesions (topography)
Although there is considerable variation from individual to individual, numerous lesions are usually present . In general, lesions of EM develop preferentially on the extremities and the face ; target lesions favor the upper extremities , as does the entire eruption of EM . The dorsal aspects of the hands and the forearms are the most frequently involved sites, but the palms, neck, face and trunk are common locations as well ( Fig. 20.2 ) . Involvement of the legs is seen less frequently. EM lesions may also appear within areas of sunburn . In addition, lesions tend to be grouped, especially on the elbows or knees .
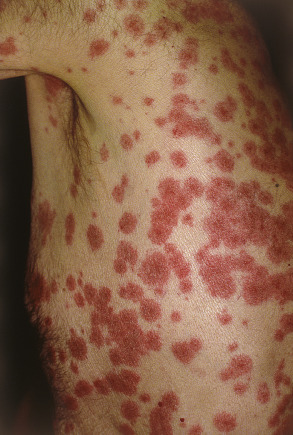
The Koebner phenomenon may be observed, with target lesions appearing within areas of cutaneous injury such as scratches (see Fig. 20.1F ) or as erythema and swelling of the proximal nail folds at sites of chronic self-trauma. The injury must precede the onset of the EM eruption because the Koebner phenomenon does not occur once the EM lesions have appeared.
Mucosal lesions
Severe mucosal involvement is characteristic of EM major. Mucosal involvement is usually absent in EM minor, and, when present, lesions are few in number and mildly symptomatic . The primary mucosal lesions of EM are vesiculobullous and rapidly develop into painful erosions that involve the buccal mucosa and lips ( Fig. 20.3 ), and less commonly the ocular and genital mucosae. On the lips, the erosions rapidly become covered by painful crusts. Erosions of the anogenital mucosa are often large and polycyclic with a moist base.
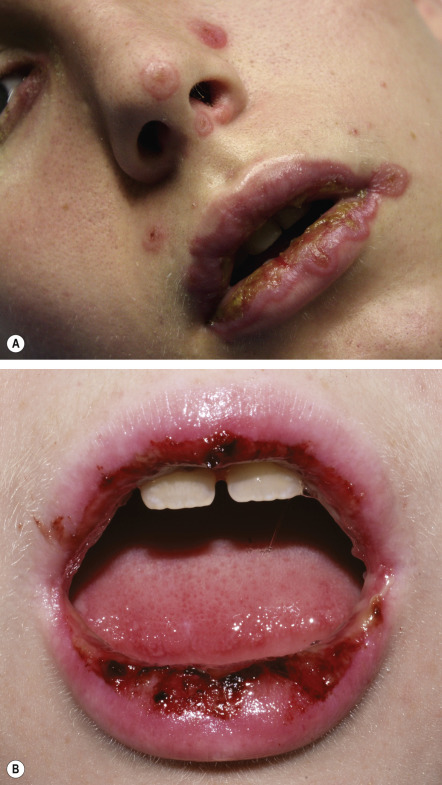
Systemic symptoms
Systemic symptoms are almost always present in EM major and absent or limited in EM minor. In EM major, the systemic symptoms that usually precede and accompany the skin lesions are fever and asthenia of varying degrees. Arthralgias with joint swelling have occasionally been described, as has pulmonary involvement resembling atypical pneumonia. Whether the latter is due to a pulmonary manifestation of EM versus an associated primary infection such as Mycoplasma pneumonia is unclear. Renal, hepatic, and hematologic abnormalities in the context of EM major are rare . By integrating these four clinical criteria, a distinction can be made between EM minor, EM major, and SJS (see Table 20.1 ).
Natural history
In EM, almost all of the lesions appear within 24 hours and full development has occurred by 72 hours . Pruritic or burning sensations within the lesions may be described. Individual lesions remain fixed at the same site for 7 days or longer .
For most individuals with EM, the episode lasts 2 weeks and heals without sequelae ; one possible rare exception is ocular damage in the setting of EM major, which may occur if adequate eye care is not promptly instituted. Occasionally, postinflammatory hyper- or hypopigmentation is seen. Patients with EM usually have an uncomplicated clinical course, although recurrences, in the case of HSV-associated EM, are quite common . One recurrence each spring, as described by von Hebra, may occur . Most individuals with recurrent HSV-associated EM have one or two episodes a year, an exception being patients receiving immunosuppressive drugs . The use of immunosuppressive agents such as oral corticosteroids may be associated with more frequent and longer episodes of EM . These individuals may have five or six episodes per year or even almost continuous disease in which one attack has not completely resolved before another occurs. The incidence of secondary bacterial infections also increases in the setting of prolonged corticosteroid use .
Pathology
EM is a clinicopathologic, not a purely histologic, diagnosis. Histologic findings are characteristic, but not specific, and are most useful for excluding entities in the differential diagnosis such as lupus erythematosus (LE) and vasculitis . In EM, the keratinocyte is the target of the inflammatory insult, with apoptosis of individual keratinocytes being the earliest pathologic finding ( Fig. 20.4 ) . As the process evolves, mild spongiosis and focal vacuolar degeneration of basal keratinocytes are observed. Superficial dermal edema and a perivascular infiltrate of lymphocytes with exocytosis into the epidermis are also seen.
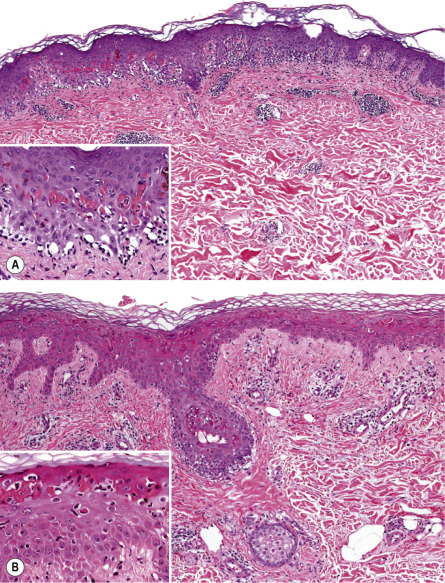
Immunofluorescence findings are nonspecific . Granular deposits of IgM and C3 around superficial blood vessels and focally at the dermal–epidermal junction have been described. Specific HSV antigens have been detected within keratinocytes by immunofluorescence, and HSV genomic DNA has been detected by PCR amplification of skin biopsy specimens .
Compared to SJS, the dermal inflammation component is more prominent in EM and the epidermal “necrolysis” component is more discrete . Large areas of full-thickness epidermal necrosis are not seen in EM.
Differential Diagnosis
Many non-dermatologists overdiagnose EM by labeling individuals with giant annular urticaria as having EM ( Fig. 20.5 ) . This is further highlighted by the term “urticaria multiforme”, which represents a variant of urticaria that is often misdiagnosed as erythema multiforme or a serum sickness-like reaction in children . This emphasizes the need to apply clinical criteria to distinguish EM from urticaria ( Table 20.3 ) . These include the presence of symmetrical fixed red papules or atypical papular target lesions, at least some of which evolve into typical target lesions. Particular attention should be given to the duration of individual lesions at a specific site and to epidermal damage in the center of the target lesions. EM papules are “fixed” at the same skin site for at least 7 days, whereas urticarial lesions last <24 hours at a particular site. The center of EM lesions demonstrates epithelial damage in the form of crusting or vesicles, whereas the center of giant urticaria is characterized by normal skin or erythema without epidermal damage. Administration of subcutaneous epinephrine (adrenaline) will clear urticaria within 20 minutes, but will not change EM lesions. Edema of the face, hands and feet can be associated with urticaria, but is usually not observed in EM .
| COMPARISON OF URTICARIA AND ERYTHEMA MULTIFORME | |
|---|---|
| Urticaria | Erythema multiforme |
| Central zone is normal skin or transient duskiness | Central zone is damaged skin (dusky, bullous or crusted) |
| Individual lesions are transient, lasting <24 hours * | Individual lesions “fixed” for at least 7 days |
| New lesions appear daily | All lesions appear within first 72 hours |
| May be associated with swelling of face, hands or feet (angioedema) | No edema |
* Can be documented by circling individual lesions with ink then observing over time.
A number of other conditions can sometimes have “target-like” lesions and mimic EM, including fixed drug eruptions, subacute cutaneous LE, Kawasaki disease, erythema annulare centrifugum, and several forms of vasculitis . A skin biopsy to exclude these entities is helpful. However, there is significant clinical and histologic overlap between EM and generalized fixed drug eruption. The total number of lesions is helpful, especially when there are hundreds, but it is also important to try to determine the number of lesions present in the initial episode (i.e. fewer lesions in fixed drug eruption). The term Mycoplasma -induced rash and mucositis (MIRM) has been proposed to describe a clinical presentation with significant mucositis (oral, ocular and anogenital) but absent to sparse cutaneous involvement in the majority of patients ; there is significant overlap with the acro-mucosal variant of EM due to Mycoplasma infection.
Recurrent EM during childhood may mimic polymorphous light eruption or juvenile spring eruption in that it may be sun-induced and develop with the first significant sun exposure in the spring . In patients with systemic LE, occasional individual lesions will mimic true target lesions of EM, but other lesions characteristic of systemic LE are usually present . That said, patients with Rowell syndrome may only have EM-like lesions as can children with Kawasaki disease. Early lesions of vasculitis, particularly urticarial vasculitis, may mimic target lesions of EM. Histologic examination assists in distinction; additional clues include an elevated ESR, autoantibodies, and low serum complement levels .
Treatment
Therapeutic options include topical and systemic treatment of the acute eruption as well as prophylactic treatment of recurrent disease. Topical therapy includes topical antiseptics for eroded skin lesions and antiseptic/antihistamine rinses and local anesthetic solutions for oral lesions. Administration of topical ophthalmic preparations should be done in conjunction with an ophthalmologist.
There are no double-blind or open trials of systemic therapies for the acute episode of EM . When a precipitating factor can be identified (e.g. HSV, M. pneumoniae ), specific therapy should be instituted; in the case of HSV, this represents chronic suppressive antiviral therapy (see below). As a rule, antiviral therapy has minimal impact if given after the appearance of the acute episode of EM. In most cases of EM, especially EM minor, symptomatic treatment will suffice. Oral antihistamines for 3 or 4 days may reduce the stinging and burning of the skin. In severe EM with functional impairment, early therapy with systemic corticosteroids (e.g. prednisone [0.5–1 mg/kg/day for 3–5 days] or pulse methylprednisolone [20 mg/kg/day for 3 days]) should be considered, despite the absence of controlled studies and the long-existing controversy regarding increasing the risk of infectious complications .
In individuals with HSV-associated EM with frequent recurrences, prophylaxis for at least 6 months with oral acyclovir (10 mg/kg/day in divided doses), valacyclovir (500–1000 mg/day, with dose depending upon frequency of recurrences), or famciclovir (250 mg twice daily) should be considered. A double-blind, placebo-controlled study in young adults with EM demonstrated the efficacy of acyclovir prophylaxis . In addition to reducing the frequency of recurrences, occasionally the beneficial effect can continue even after the antiviral drug is discontinued. In non-responsive patients, the dose of the medication may be doubled, or a substitution made of the antiviral drug. However, as stated previously, treatment with acyclovir or valacyclovir after symptoms appear is ineffective . Obviously, in those patients in whom EM is precipitated by factors other than reactivation of HSV, antivirals will be of no benefit.
In patients with recurrent EM that is resistant to prophylactic antiviral therapy, especially severe cases, several therapeutic approaches have been described; however, none have been validated by controlled trials and none have been consistently effective or free of side effects. Among these are: azathioprine (100 mg/day for several months), prednisone (0.5 mg/kg/day for several months), thalidomide, dapsone, cyclosporine, mycophenolate mofetil, and PUVA .
Stevens–Johnson Syndrome and Toxic Epidermal Necrolysis
▪ Toxic epidermal necrolysis – Lyell syndrome
- ▪
Prodrome of upper respiratory tract symptoms, fever, and painful skin
- ▪
Stevens–Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are two rare, potentially fatal, adverse cutaneous drug reactions of differing severity, characterized by mucocutaneous tenderness and erythema as well as extensive exfoliation
- ▪
SJS is characterized by <10% body surface area of epidermal detachment, SJS/TEN overlap by 10–30%, and TEN by >30%
- ▪
The medications most frequently incriminated are allopurinol, nonsteroidal anti-inflammatory drugs, antibiotics, and aromatic anticonvulsants; TEN and SJS usually occur 7–21 days after initiation of the responsible drug
- ▪
The average mortality rate is 1–5% for SJS and 25–35% for TEN; the mortality rates are often higher in the elderly and those with a very large surface area of epidermal detachment
- ▪
Exfoliation is due to extensive death of keratinocytes via apoptosis; the latter is mediated via the cytotoxic secretory protein granulysin and interaction of the death receptor–ligand pair Fas–FasL
- ▪
Optimal medical management of SJS and TEN requires early diagnosis, immediate discontinuation of the causative drug(s), and rapid initiation of supportive care
- ▪
Additional therapies include IVIg, cyclosporine, pulse corticosteroids, and targeted immunomodulators, but to date none have proven efficacy based upon prospective controlled trials
Introduction
Stevens–Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) are rare, acute and life-threatening mucocutaneous diseases that are nearly always drug-related. They are a consequence of extensive keratinocyte cell death that results in the separation of significant areas of skin at the dermal–epidermal junction, producing the appearance of scalded skin. This extensive cell death also results in mucous membrane detachment, and it contributes to the characteristic symptoms of SJS and TEN, which include: high fever, moderate to severe skin pain, anxiety, and asthenia. The disease runs an unpredictable course. An initially benign-appearing dermatosis can progress rapidly, and once overt skin detachment has occurred, it is difficult to determine when it will end. Several studies have addressed the clinical signs of SJS and TEN, and there is now a framework of diagnostic criteria. Mortality risk can be calculated by applying a severity-of-illness score specifically developed for predicting the clinical outcome of TEN (SCORTEN; see below).
As prognosis is correlated with the speed at which the culprit drug is identified and withdrawn, it is crucial to establish the correct clinical diagnosis rapidly, so the causal drug(s) can be discontinued and appropriate medical treatment begun as soon as possible. Although TEN, and especially SJS, were historically considered to be part of a spectrum of diseases that included EM major (all of which have clinically similar mucosal lesions), these disorders should now be distinguished, as their cause, management, and/or prognosis are usually distinct (see Table 20.1 ). SJS and TEN should also be distinguished from dermatoses such as staphylococcal scalded skin syndrome, generalized fixed drug eruption, drug-induced linear IgA bullous dermatosis, toxic erythema of chemotherapy, and acute generalized exanthematous pustulosis, as their management and prognosis are also quite different.
High-quality supportive treatment, ideally in intensive care units with modern equipment and trained nursing staff, is the current standard of care and can improve outcome. No specific therapy for SJS and TEN has yet shown efficacy in prospective, controlled clinical studies, in part because the low incidence of SJS and TEN plus their life-threatening potential make randomized clinical trials difficult to perform. Despite this, conceptually interesting approaches based upon known elements of the pathogenesis of SJS and TEN and small case series have been described (see below).
History
In 1922, two US physicians, Stevens and Johnson, described an acute mucocutaneous syndrome in two young boys. The condition was characterized by severe purulent conjunctivitis, severe stomatitis with extensive mucosal necrosis, and “EM-like” cutaneous lesions. It became known as Stevens–Johnson syndrome (SJS) and was recognized as a severe mucocutaneous disease with a prolonged course and occasional fatalities . SJS was later designated as EM major by Bernard Thomas in 1950 . However, recent clinical investigations have made it clear that the term “EM major” should not be used to describe SJS as they are distinct disorders (see Table 20.1 ) .
In 1956, Alan Lyell described four patients with an eruption “resembling scalding of the skin objectively and subjectively”, which he called “toxic epidermal necrolysis”. “Toxic” referred to toxemia – circulation of a toxin – which was thought to be responsible for the constitutional symptoms and epidermal necrosis. Lyell coined the term “necrolysis” by combining the key clinical feature “epidermolysis” with the characteristic histopathologic feature “necrosis”. He also described an attack on the mucous membranes as part of the syndrome, and noted that there was very little inflammation in the dermis, a feature that was later referred to as “dermal silence” . This feature contrasted with the obvious inflammatory infiltrate of other blistering disorders such as EM, dermatitis herpetiformis, and bullous pemphigoid. TEN was considered at the time to be a cutaneous reaction pattern to multiple stimuli, including drugs (e.g. sulfonamides) and microbes (e.g. Staphylococcus ) .
The description of a subgranular epidermal split in neonatal mice following exposure to phage group II S. aureus , the subsequent discovery of a new staphylococcal exotoxin called epidermolytic toxin, and the absence of overt keratinocyte necrosis led to the distinction between TEN and what would come to be known as “staphylococcal scalded skin syndrome” (SSSS; see Ch. 74 ) . It should be noted here that one of Lyell’s original patients actually represented SSSS, and although by that time he had already noted the histologic differences between TEN and SSSS, they were attributed to different degrees of injury.
As more patients with TEN were reported in the years following Lyell’s treatise, it became clear that certain drugs such as sulfonamides, pyrazolones (e.g. phenylbutazone), barbiturates, and aromatic anticonvulsants were principally associated with TEN. At the same time, drugs were increasingly being incriminated as a cause of EM associated with severe stomatitis. Hence, EM epidermal type (as defined by Orfanos et al. in the mid 1970s), SJS and TEN were, at the time, considered to be part of a continuous spectrum of cutaneous reactions. It was clear, however, that HSV was the major cause of EM, and that this virus was not associated with cases of TEN. In the late 1990s, the group of Jean-Claude Roujeau clarified this issue by providing clinical evidence that EM and SJS were clinically distinct disorders with different causes and prognoses . Increasingly, SJS and TEN are considered to be two ends of a spectrum of severe epidermolytic adverse cutaneous drug reactions, differing only by the extent of skin detachment (see Table 20.1 ).
Epidemiology
SJS and TEN are rare diseases that affect women more often than men. Certain patient groups have an increased risk of developing SJS/TEN, including those who metabolize drugs at a decreased rate (slow acetylator genotypes), are immunocompromised (e.g. HIV infection), are undergoing radiotherapy while concomitantly receiving anticonvulsants (see Fig. 139.10 ), or have specific human leukocyte antigen (HLA) alleles ( Table 20.4 ). Examples of the latter are HLA-B*15:02 in Asians and East Indians who are exposed to carbamazepine and HLA-B*58:01 in Han Chinese exposed to allopurinol. Consequently, the FDA recently recommended genotyping of all Asians for the allele HLA-B*15:02 prior to the administration of carbamazepine. In individuals with AIDS, the risk of developing TEN is 1000-fold higher than in the general population .
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


