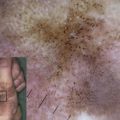
Epidermolysis bullosa (EB) with pyloric atresia (PA) is a rare form of EB. This article describes the clinical and pathologic features and molecular genetics of EB-PA, the mutations in the α 6 β 4 integrin and plectin genes that cause EB-PA, and the clinical implications of molecular genetics on EB-PA.
Clinical and genetic heterogeneity of EB
Epidermolysis bullosa (EB) is a heterogeneous group of skin fragility syndromes with the diagnostic hallmark of blistering and erosions of the skin. Proper diagnosis and subclassification of different forms of EB can be challenging for general practitioners because of considerable phenotypic variability, as reflected by the complex classification schemes riddled with eponyms. The most streamlined classification divides EB into 3 broad categories depending on the precise location of tissue separation within the cutaneous basement membrane zone, as determined by diagnostic transmission electron microscopy or by immunoepitope mapping: (1) in the simplex forms (EBS), tissue separation takes place within the basal keratinocytes; (2) the junctional forms (JEB) show blistering within the dermal-epidermal basement membrane, frequently within the lamina lucida; and (3) in the dystrophic forms (DEB), tissue separation is below the lamina densa within the upper papillary dermis at the level of anchoring fibrils. The inheritance of different forms of EB is either autosomal dominant or autosomal recessive. It is now known that mutations in 10 different genes expressed within the cutaneous basement membrane zone underlie the classic simplex, junctional, and dystrophic forms of EB. The types and combinations of mutations, their consequences at the mRNA and protein levels, when placed in the context of the individuals’ genetic background, including modifier genes, and the environmental trauma, all contribute to the severity of the disease, explaining the phenotypic variability in this group of disorders.
In addition to skin involvement, different forms of EB can be associated with extracutaneous manifestations; these include hair, nail, and tooth abnormalities, ocular findings, and fragility of the epithelia in upper respiratory, urogenital, and gastrointestinal tracts. There are 2 rare forms of EB, one associated with late-onset muscular dystrophy (EB-MD), and another one with congenital pyloric atresia (EB-PA). EB-MD has been shown to result from mutations in the plectin gene ( PLEC1 ), which encodes a large, approximately 500-kDa adhesion molecule. In the skin, the binding partners of plectin include basal cell keratins (KRT5 and KRT14), α 6 β 4 integrins, and type XVII collagen/the 180-kDa bullous pemphigoid antigen, thus serving as a bridge between the intermediate filament cytoskeleton and hemidesmosomes within the basal keratinocytes ( Fig. 1 ). In addition to skin, plectin is expressed in various tissues, including striated muscle and gastrointestinal epithelia. Specifically, in skeletal muscle, plectin is expressed in the sarcolemma and the Z-lines, thus participating in the formation of the intermyofibrillar-desmin cytoskeleton. Consequently, expression of plectin in the skin and in the skeletal muscle explains the consequences of mutations in 2 different organ systems in EB-MD, characterized by skin blistering and muscular dystrophy.
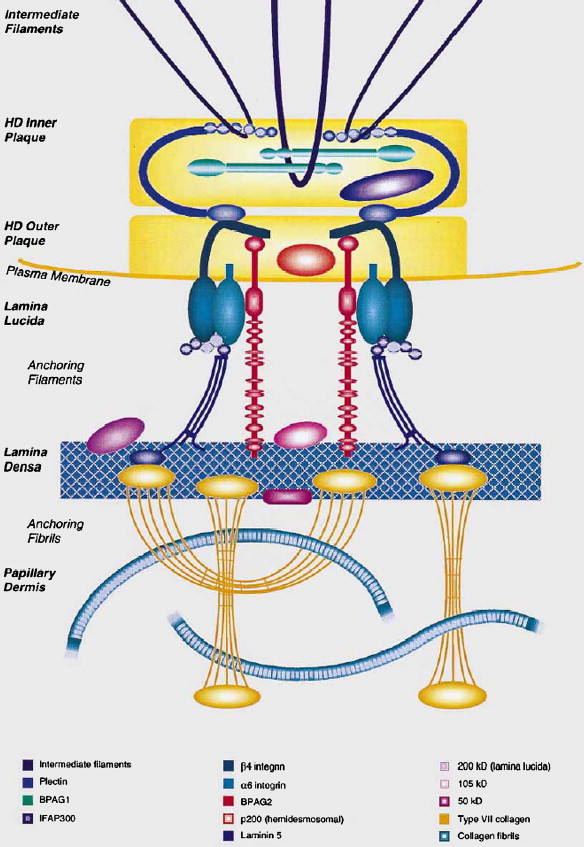
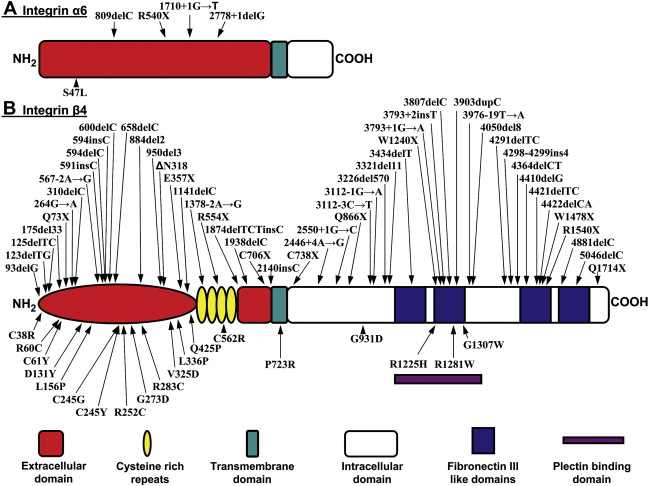
Another gene/protein system harboring mutations in patients with EB-PA is the α 6 β 4 integrin; the corresponding subunit polypeptides of this hemidesmosomal protein are encoded by the ITGA6 and ITGB4 genes. This transmembrane protein is an integral part of hemidesmosomes serving a structural role in the attachment of the basal keratinocytes to the underlying basement membrane, and serving as signaling molecules. Within the intracellular milieu of basal keratinocytes, α 6 β 4 is linked to the cytokeratin network by plectin; the α 6 β 4 -plectin interaction is crucial for hemidesmosome stability, and it has been proposed that this association acts as an initiation step in the assembly of hemidesmosomes. The α 6 β 4 integrin is characteristically expressed in various epithelial tissues, including human skin and the gastrointestinal tract, in which it functions as a receptor for laminin-332 (laminin-5), a major extracellular component of the epidermal basement membrane (see Fig. 1 ). The subunit responsible for intracellular interactions of α 6 β 4 , including binding to plectin, is the cytoplasmic domain of the β 4 subunit, which is unusually large (approximately 1000 amino acid residues) and shares little similarity with other integrin β-subunits. The β 4 cytodomain has a modular organization with 4 fibronectin type III (FnIII) domains arranged in 2 pairs of tandem repeats separated by a connecting segment region ( Fig. 2 ). The N-terminal region of plectin interacts with the β 4 subunit at multiple sites. The primary contact is established between the actin binding domain of plectin and the first pair of FnIII domains and a small region of the connecting segment of β 4 polypeptide ( Figs. 2 and 3 ). In the extracellular domain, β 4 integrin has a segment of cysteine-rich repeats.