TABLE 23-1. Key Features of the Major Epidermal Nevus Syndromes | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
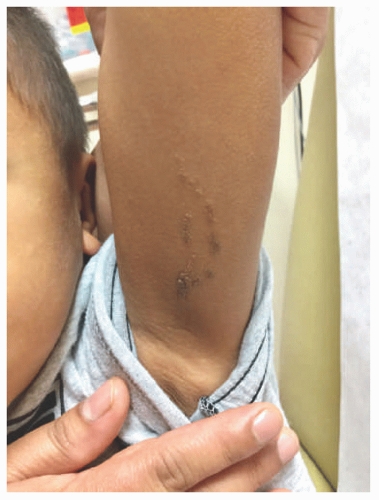 FIGURE 23-1. An epidermal nevus on the inner aspect of the arm presenting as a linear plaque formed by confluent fleshy papules. |
consider as a manifestation of segmental or zosteriform Darier disease.12,13,14 Rare cases display a distinctive pattern of rectangular elongation of the rete ridges with palisading of basal keratinocytes and are referred to as papular epidermal nevus with skyline basal cell layer (PENS).15 These are associated with extracutaneous findings (neurologic, in particular) in approximately 50% of cases.16
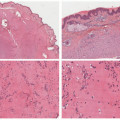
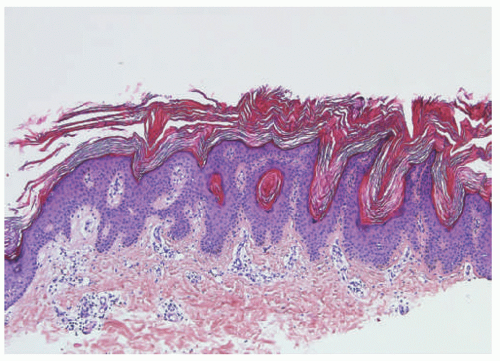 FIGURE 23-2. Epidermal nevus frequently resembles seborrheic keratosis histologically, with orthokeratotic hyperkeratosis, papillomatosis, and epidermal acanthosis. |
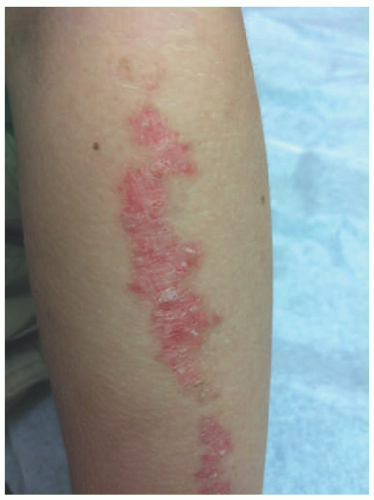 FIGURE 23-3. Inflammatory linear verrucous epidermal nevus (ILVEN) presents as an erythematous and scaly linear plaque, resembling linear psoriasis. |
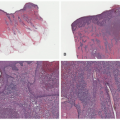
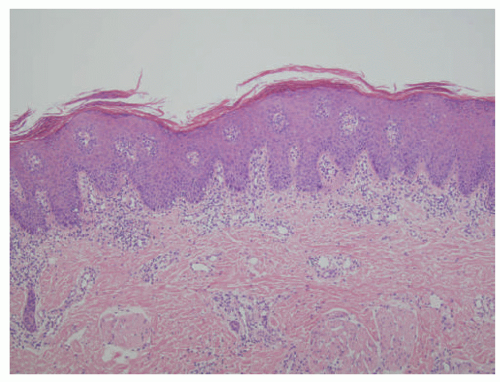 FIGURE 23-4. Horizontal alternation of orthokeratotic and parakeratotic hyperkeratosis is characteristic of inflammatory linear verrucous epidermal nevus (ILVEN), although the histologic features can be difficult to separate from psoriasis. |
plaques that primarily involves skin folds. The posterior and lateral folds of the neck are commonly affected. Early acanthosis nigricans may be heralded by slight hyperpigmentation that later eventuates into thickening of the skin. Other affected areas include the dorsal knuckles, genitalia, perineum, face, thighs, breast, and axillae. Generalized acanthosis nigricans may occur, which can present with involvement of the oral mucosa, lips, and eyelids and may herald an underlying malignancy.33 Similarly, the finding of “tripe palms,” or velvety thickening of the palms and dorsal hand, may also be paraneoplastic.34 Rarely, acanthosis nigricans may present in the form of an epidermal nevus, which may be heritable in autosomal dominant fashion and can present at birth, childhood, or puberty.35
TABLE 23-2. Conditions Associated With Acanthosis Nigricans | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
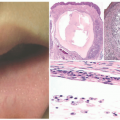
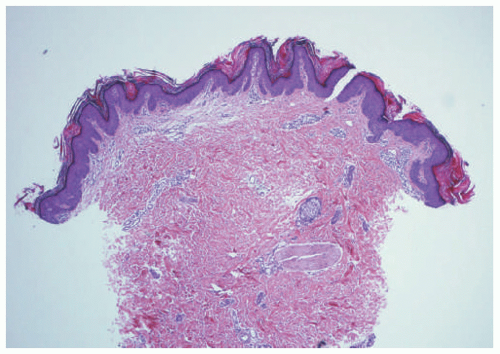 FIGURE 23-5. Acanthosis nigricans is characterized by hyperkeratosis and papillomatosis. The appearances are similar to those of confluent and reticulated papillomatosis (CARP) and some cases of epidermal nevus. |
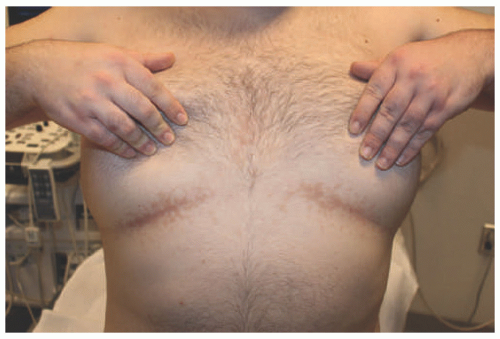 FIGURE 23-6. Confluent and reticulated papillomatosis (CARP) frequently presents in adolescent patients as hyperpigmented papules in the inframammary region, which coalesce to form thin reticulate plaques. |
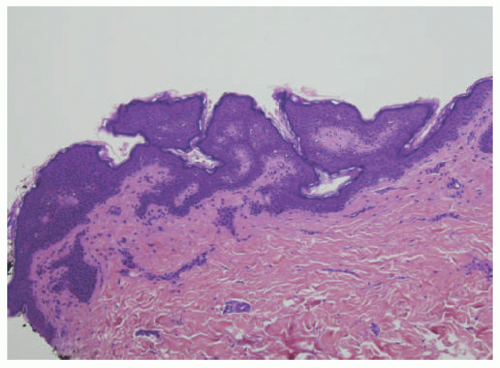 FIGURE 23-7. The histologic features of confluent and reticulated papillomatosis (CARP) include orthokeratotic hyperkeratosis and slight papillomatosis. Fungal yeast forms are often identifiable on the surface. |
of its target Smoothened (SMO, 7q32). Germline mutations in PTCH1 are associated with autosomal dominantly inherited basal cell nevus syndrome (BCNS).55,56 Mutations in the gene encoding tumor suppressor p53 (17p13.1) as well as CDKN2A (9p21) may also play a role. Recent data suggest that a single-nucleotide polymorphism in mismatch repair genes MSH2 (2p) and MLH1 (Chr. 3) as well as variants in the melanocortin 1 receptor (MC1R, 16q) gene may predispose individuals to BCC.57,58 A novel hereditary BCC syndrome, wherein patients develop multiple hereditary infundibulocystic variant BCCs, has recently been attributed to heterozygous splice site mutations in one copy of the tumor suppressor known as suppressor of fused gene (SUFU, 10q), a component of the sonic hedgehog pathway.59,60
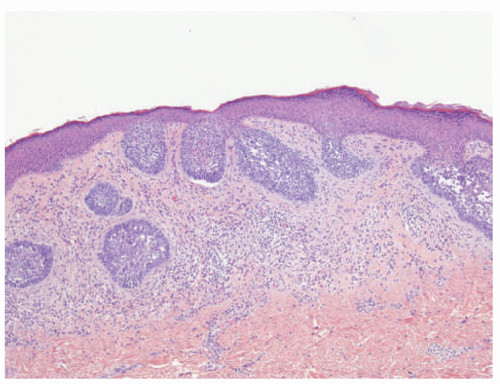 FIGURE 23-8. Basal cell carcinoma in children shows similar features to those seen in adults, although background solar elastosis is typically absent. This sporadic lesion arose on the shoulder of a 14-year-old patient. |
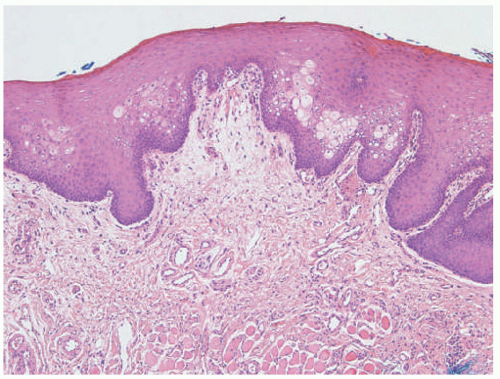 FIGURE 23-9. White sponge nevus displays parakeratosis, acanthosis, and prominent clearing of suprabasal keratinocytes. |
mucosa.92,93,94,95,96 A single, round to oval, sharply circumscribed, yellow to flesh-colored, pebbly thin plaque is often noted at birth and slowly thickens during childhood, while also assuming a more yellow-orange hue. Around adolescence, as a result of hormonal stimulation of lesional sebaceous glands, a more prominent thickening occurs, leaving a verrucous to cerebriform waxy smooth surface. Behind the ear, a linear configuration is often noted. Importantly, multiple lesions accompanied by seizures; intellectual impairment; and ocular, craniofacial, or skeletal abnormalities should raise suspicion for Schimmelpenning syndrome. The additional presence of a speckled lentiginous nevus along with skeletal and neurologic defects should raise concern for phakomatosis pigmentokeratotica.
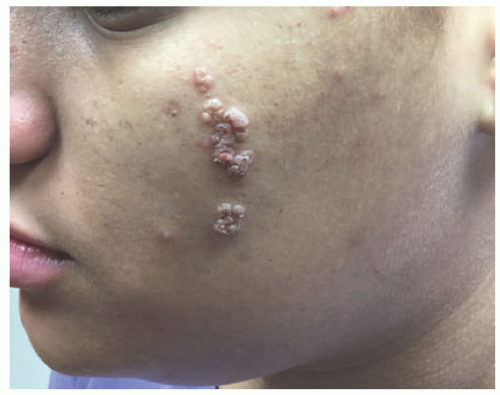 FIGURE 23-10. Nevus sebaceus presenting as a plaque with a pebbled appearance on the face of an adolescent patient. |
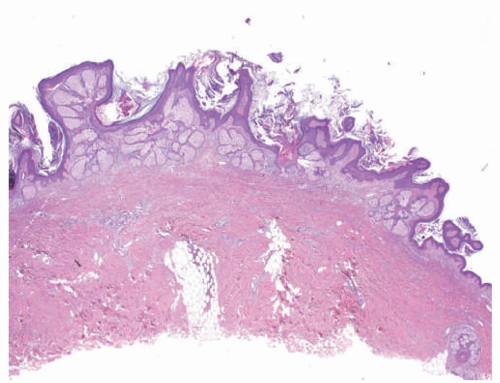 FIGURE 23-11. Nevus sebaceus displaying papillomatous epidermal hyperplasia and abnormal sebaceous glands. Note the absence of terminal hairs in this biopsy from the scalp, which can be a useful diagnostic clue. |
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


