22 Embryology of the craniofacial complex
Synopsis








What is unique about craniofacial development?
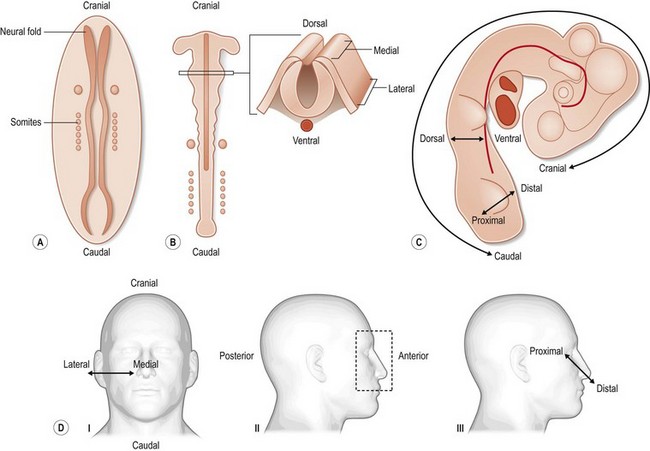
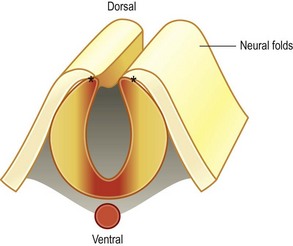
In fact, there are a number of unique features that clearly distinguish craniofacial development from the development of other tissues in the body. One of these unique features is the dual origin of craniofacial tissues: the skeletal scaffold and most of the connective tissues, including blood vessels, originate from a group of cells called the cranial neural crest, whereas the musculature and some parts of the skull originate from mesoderm. A second unique feature is the cadre of complex, reciprocal tissue–tissue interactions between the neuroectoderm, the mesenchyme, and facial ectoderm that drive normal development. A third novel feature is the elaborately choreographed morphogenetic movements – caused by both passive cell displacement and active cell migration – that define head development (Fig. 22.1). An example of active cell migration is movement of cranial neural crest cells from the dorsal neural folds into the branchial arches and more ventral positions in the facial prominences (Fig. 22.2). Any process that disrupts the rate, the timing, or the extent of these complex cellular behaviors can result in a craniofacial birth defect. In the following paragraphs, a brief overview of these early cellular movements, and the molecular pathways governing those movements, will be discussed.
The initiation of craniofacial development
Gastrulation and the establishment of the craniocaudal and mediolateral axes
In the 3rd week of human development, the embryoblast grows, it divides into two, then three, layers. This process, called gastrulation, ultimately results in the formation of three primary germ layers (ectoderm, mesoderm, and endoderm). Even before this trilaminar disc of embryonic cells is established, however, it is apparent that the cranial (also known as rostral, or anterior) region of the embryo is distinct from the caudal (posterior) region (Fig. 22.1). At the caudal end of the embryo, a group of cells known as the “primitive streak” emerges from the “primitive node.” This epiblast-derived column of cells (chorda-mesoderm) progresses cranially via convergent extension movement until it reaches the prechordal plate, which is the site of the future mouth. The first cells to move anteriorly give rise to the pharyngeal endoderm and head mesenchyme.
By the end of the 4th and beginning of the 5th gestational week of human development, the craniocaudal (head to tail) axis is established. The mediolateral axis is also specified at this juncture, with “medial” denoting the midline of the embryo and “lateral” signifying the borders of the embryo (Fig. 22.1). The molecular specification of the craniocaudal, mediolateral, and dorsoventral axis in vertebrates is obviously quite complex and is described in detail elsewhere.1–3 For this review, we will limit our discussion to those molecular pathways that, when disrupted, produce the more common craniofacial malformations.
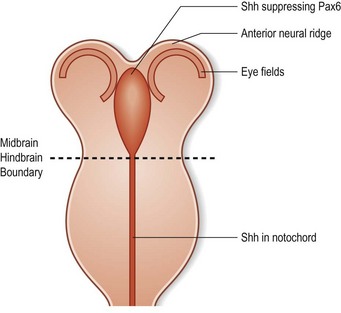
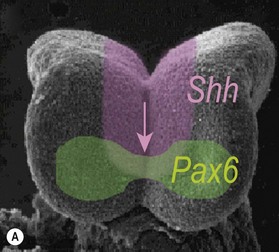
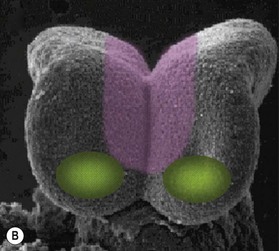
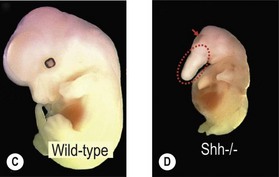
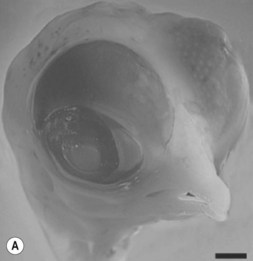
One of the earliest molecules that define the mediolateral axis is part of the “Hedgehog” family of secreted growth factors. Sonic hedgehog (shh), its receptor Patched (ptch), and at least one of its downstream targets, the transcription factor Gli1, are expressed in the medial region of the neural plate (Fig. 22.3). In this midline domain, Shh signaling actively represses Pax6, a transcription factor that is required for the specification of the presumptive eye field in the anterior neural plate. Shh signaling normally represses Pax6 expression in the midline of the presumptive eye field and in doing so, bifurcates into two, symmetrical regions (Fig. 22.4). By this mechanism, a single eye field develops into two, separate eye fields. When Shh signaling is lost or disrupted at this stage of embryonic development, Pax6 expression persists in the midline and as a consequence, only a single eye field is evident (Fig. 22.5). Therefore, a loss of Hedgehog function at this early stage of development produces embryos with cyclopia (discussed below). This is a highly conserved pathway between various species and is present in all vertebrates (Fig. 22.6). A single median eye is usually associated with an absence of the optic stalks, the optic chiasm, and the pituitary gland.4 The presence of a single eye is also accompanied by a proboscis, a medial tubular appendage lacking a nasal septum.
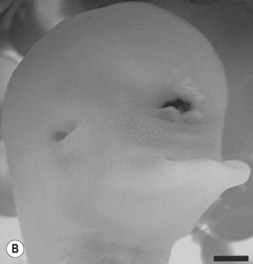
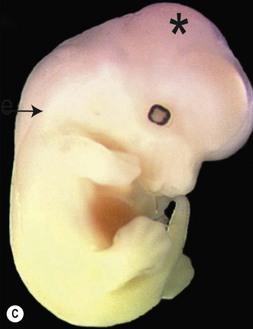
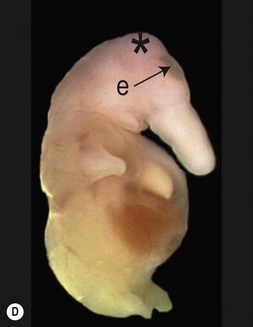

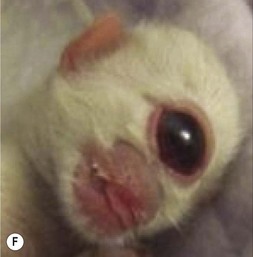
Disruptions in mediolateral patterning produce severe HPE phenotypes
Cyclopia is the most severe manifestation of the malformation sequence known as “holoprosencephaly” (HPE). HPE is the most common structural malformation of the forebrain in humans, occurring in approximately 1 : 10–20 000 live births.5 In addition to cyclopia, HPE is associated with other craniofacial anomalies, including cebocephaly, ethmocephaly, and median cleft lip. Milder HPE phenotypes are known as microforms and they include microcephaly, microphthalmia, ocular hypotelorism, midfacial hypoplasia, and cleft lip with or without cleft palate.6 In perhaps the mildest HPE phenotype, affected individuals exhibit only subtle midline abnormalities such as presence of a single upper central incisor.6
The etiology of HPE is as heterogeneous as its phenotypic presentation (Table 22.1). Despite this, almost all of the gene mutations identified to date lead to a disruption in Hedgehog signaling.
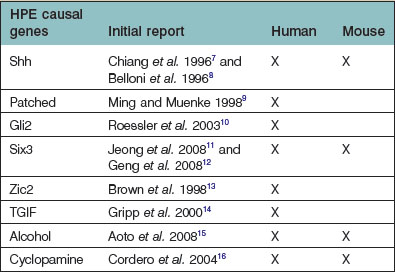
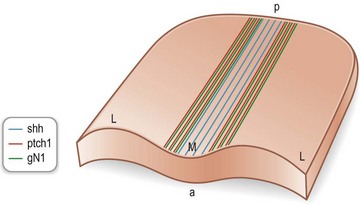
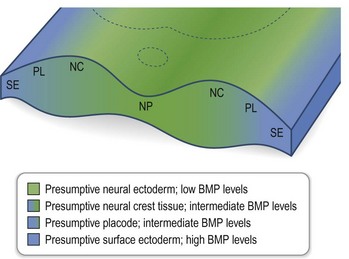
Simultaneous with specification of the medial axis is patterning of the lateral regions of the anterior neural plate. Lateral plate patterning is regulated in large part, by members of the bone morphogenetic protein (BMP) family, which are expressed by cells located at the boundary between the neural and non-neural ectoderm (Fig. 22.7). Signaling by BMP, plays an important role in the early stages of head formation and it is hypothesized that BMP activity promotes non-neural ectoderm while it inhibits neural ectoderm. This hypothesis is supported by mice with mutations in the extracellular antagonists that attenuate BMP, such as Chordin and Noggin, which display phenotypes remarkably analogous to human HPE sequence.17
Neurulation and the generation of the neural crest
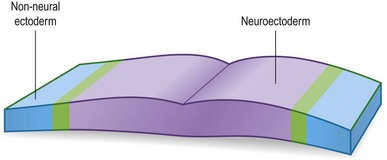
During the process of neurulation, the neural plate “rolls up” to form the neural tube and in effect, this morphogenetic movement subdivides the ectoderm into two distinct tissues: the neural ectoderm, which will form the brain and spinal cord, and the non-neural (i.e., surface) ectoderm that will cover the embryo and later give rise to the epidermis (Fig. 22.8).
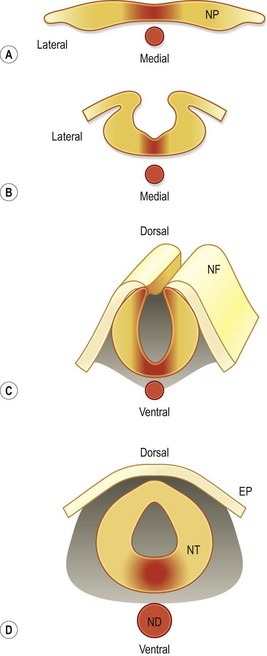
In considering how the craniofacial tissues achieve their ultimate arrangement, it is helpful to remember that those regions of the neural plate that were once positioned in the center (medial region) eventually become positioned in the ventral part of the neural tube, and those regions that were lateral at the neural plate stage eventually become located dorsally in the neural tube (Fig. 22.8). The reason that this repositioning is so important is that gene products, whose function it is to specify the midline of the neural plate, end-up having important roles in patterning the ventral neural tube. Likewise, molecules that regulate patterning in the lateral neural plate eventually have critical roles in dorsal neural tube specification. Two examples are Shh in the medial neural plate/ventral neural tube (Fig. 22.3) and BMPs in the lateral neural plate and dorsal neural tube (Fig. 22.9).
Just prior to fusion of the neural folds, a population of cells emerges from the junction of the neural and non-neural ectoderm. This population of cells is known as the neural crest, and their role in head development is essential. In the head region, neural crest cells give rise to the majority of the neural, odontogenic, and skeletogenic tissues of the head, as well as melanocytes, some intrinsic eye muscles, pericytes, which encase blood vessels in the head, and adipocytes In other parts of the body, neural crest cells yield neurons and glial cells of the entire peripheral nervous system and also endocrine cells.18 The astounding amount of neural crest derivatives, coupled with their early embryonic appearance, have led some to refer to neural crest cells as the fourth germ layer.
Neural crest cells are derived from ectoderm, but through a carefully orchestrated process, they detach and acquire a mesenchymal (i.e., migratory) fate. This epithelial to mesenchymal transition (EMT) is a unique feature of normal neural crest cells, but is also seen in metastatic cells.19 The highly invasive behavior of normal neural crest cells closely parallels cancerous cells that fail to respect tissue boundaries. As a consequence, considerable effort has gone into understanding the molecular regulation of neural crest cell formation.
Migration of the cranial neural crest into the facial prominences
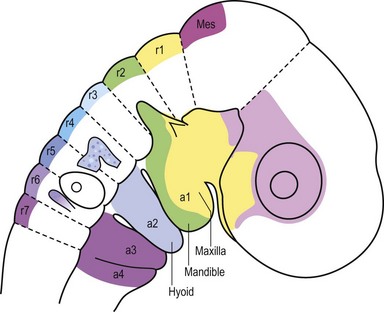
In the head region, neural crest cell migration begins at the dorsal neural folds (Fig. 22.10). Neural crest cells migrate from the dorsal neural tube into the prominences of the face; and, in avians, this migratory stream is divided into three components: those neural crest originating from rhombomere 2 migrate into the first arch; those arising from rhombomere 4 migrate into the second arch; and those neural crest cells from rhombomere 6 migrate into the third arch (Fig. 22.2).20 This migratory route appears to be largely the same in mammals and avians, and thus the majority of our detailed understanding of neural crest cell behavior has come from elegant experiments conducted using chick/quail chimeras by Nicole Le Douarin and colleagues.21,22 Now, new imaging strategies can visualize, at the single cell level, the emigration of the cranial neural crest into the facial prominences, and these studies in large part, verify the earlier experiments.
Cranial neural crest mesenchymal cells migrate extensively throughout the embryo, most notably into the facial prominences. How do neural crest cells know which pathway to take, and where to end-up? New data indicate that this population of cells uses “cues” found on adjacent epithelial cells to direct their migration. This process is exemplified by the reciprocal signaling between cells expressing the Ephrins and cells expressing the Eph receptors, semaphorin III/collapsin I. Ephrins and Eph receptors are membrane bound proteins that function as a ligand-receptor pair.23 Upon ligand-receptor binding, a molecular conduit for cross-talk is established between neural crest cells and adjacent tissues.24,25 This was determined in part by experiments where truncated Ephrin receptors, which acted in a dominant negative fashion, were over-expressed in embryos. As a result of over-expression, neural crest cells failed to migrate appropriately, which indicated that Ephrin/Eph signaling was crucial for the proper migration of these cells. Subsequent experiments in avians indicate that Ephrin/Ephs act as bifunctional guidance cues, responsible for repelling some migrating neural crest cells and stimulating the migration of other cell types.26 A more detailed understanding of Ephrin/Eph signaling will undoubtedly elucidate further how cranial neural crest migration is orchestrated during craniofacial development.
Establishment and fusion of the facial prominences
The basic morphology of the face is established between the 4th and 10th weeks of human development. The face is formed as a result of fusion of the midline frontonasal prominence, and three paired prominences, the maxillary, lateral nasal, and mandibular prominences (Fig. 22.11). Each of these prominences is filled with cranial neural crest cells that originated at different positions along the neural tube (Fig. 22.2). The derivatives of each of the prominences will be discussed separately.
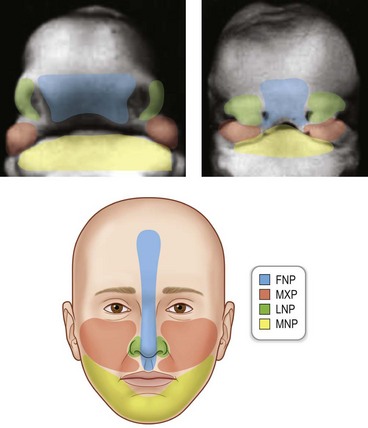
The frontonasal prominence
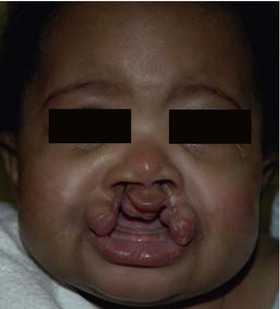
The frontonasal prominence gives rise to the forehead, midline of the nose, the philtrum, the middle portion of the upper lip, and the primary palate. Interruptions in frontonasal growth often result in a bilateral cleft lip, where the primary palate frequently “everts” (Fig. 22.12). In the mildest cases, clefts involving frontonasal prominence-derived structures may be limited to a notch in the vermillion border of the lip. In more severe cases, frontonasal clefts involve all of the tissues of the lip and these cases may most likely occur because of a failure of fusion between the frontonasal and maxillary prominences.
The lateral nasal prominences
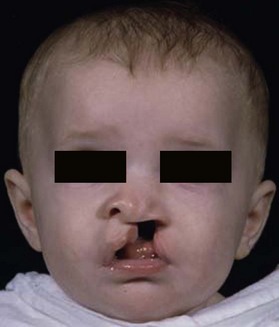
The lateral nasal prominences give rise to the alae of the nose (Fig. 22.11). Clefts that involve the side of the nose often result from a failure in the fusion between the lateral nasal prominences and either the frontonasal or the maxillary processes (Fig. 22.13).
The maxillary prominences
The maxillary prominences give rise to the upper jaw and the sides of the face, the sides of the upper lip, and the secondary palate. The secondary palate separates the nasal passage from the pharynx, and this bony structure is derived from cranial neural crest cells that populate the maxillary prominences. In a stepwise manner, the palatal shelves extend vertically on either side of the tongue, and then subsequently rotate to a horizontal plane dorsal to the tongue, and fuse (Fig. 22.14). Initially, the palatal shelves are lined by an epithelium that, just prior to fusion, is partially sloughed off, leaving only the basal epithelial layer to cover the palatal shelves. This basal epithelial layer is known as the medial edge epithelium (MEE) and as the shelves grow towards the midline, the MEE of each shelf approximates and forms the midline epithelial seam (MES). The MES is then removed through the process of mesenchymal to epithelial transition (MET, the opposite of EMT), which leads to a confluence between the mesenchyme of the palatal shelves. Perturbations caused by genetic, mechanical, or teratogenic factors can occur at any of these steps, and frequently result in a cleft of secondary palate. Another type of secondary palatal clefting is palatal insufficiency, which is often caused by inadequate outgrowth of the maxillary prominences (Fig. 22.15).

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


