Ectodermal Dysplasias: Introduction
|
The ectodermal dysplasias (EDs) are a group of inherited disorders that share in common developmental defects involving at least two of the major structures classically held to derive from the embryonic ectoderm—hair, teeth, nails, and sweat glands. Freire-Maia and Pinheiro1 published an exhaustive review and classification system for these disorders using a numeric system of 1 (hair), 2 (teeth), 3 (nail), and 4 (sweat glands) for characterization. This system has little use in practice, although it did allow for a rational approach to a previously chaotic field. Freire-Maia and Pinheiro laid claim to more than 150 ectodermal disorders in their most recent compilation.2 Several other classification schemes have been suggested. Each has limited clinical application but can help in ordering thinking about these disorders.3,4 In an attempt to move to a more useful and unified system, an international classification conference was convened in 2008 to initiate this process which is still ongoing.5
This chapter covers only the more common of these conditions and those in which ectodermal defects are likely to bring them to a dermatologist for diagnosis and medical attention. Fig. 142-1 is an algorithm showing the approach to the diagnosis of EDs. The first step in the algorithm for making a specific diagnosis of an ED is to determine the presence of sweating (hidrotic) or absence of sweating (hypohidrotic/anhidrotic). The involvement of other ectodermal structures and of nonectodermally derived tissues provides further branching points in a diagnostic hierarchy. Mode of inheritance may differ within a seemingly uniform diagnostic group, and care must be taken in evaluating family members before providing recurrence risks.
Figure 142-1

Approach to the diagnosis of ectodermal dysplasias. AD = autosomal dominant; ADULT = acro-dermato-ungual-lacrimal-tooth syndrome; AEC = ankyloblepharon filiforme adnatum–ectodermal dysplasia–cleft palate syndrome; AR = autosomal recessive; ED = ectodermal dysplasia; EEC = ectrodactyly–ED–cleft lip/palate syndrome; HED = hypohidrotic ectodermal dysplasia; IM = immune defects; X-LR = X-linked recessive.
Within the last few years, causal genes have been identified for many of the EDs. Given continuing discoveries, the reader is directed to the following resources: http://proxy.library.upenn.edu:8489/OMIM (Online Catalog of Mendelian Inheritance in Humans) and (an up-to-date listing of laboratories offering molecular testing). The National Foundation for Ectodermal Dysplasias (http://www.nfed.org) is a lay support group that has numerous informative pamphlets for families and physicians, as well as a strong advocacy program for dental care, insurance coverage, and research.
Hypohidrotic Ectodermal Dysplasia [Anhidrotic Ectodermal Dysplasia, Christ–Siemens–Touraine Syndrome; OMIM #305100]
One of the earliest descriptions of hypohidrotic ectodermal dysplasia involving male first cousins and their grandmother with sparse hair, missing teeth and dry skin was described in 1848 by Thurnam.6 Darwin described this condition in his writings from 1875.
X-linked hypohidrotic ED (XLHED) occurs in all racial groups and is thought to have an incidence at birth of anywhere from 1 in 5,000–17,000 births.7 Accurate prevalence and incidence data are not available.
XLHED results from alterations in the gene ectodysplasin (EDA, EDA1, HED) located at Xq12-13.8 It codes for a transmembrane protein, ectodysplasin, which is composed of 391 amino acids and has alternative splicing forms, the significance of which is not known. A multitude of mutations in this gene causing XLHED have been identified. There does not appear to be a correlation between the nature of the mutation and the clinical features9 (i.e., to date, there have been no phenotype–genotype correlations). Interfamilial and intrafamilial variation occurs to a mild degree. Ectodysplasin belongs to the tumor necrosis factor family and plays a role in regulation of the formation of ectodermal structures. It forms trimers and is expressed in keratinocytes, the outer root sheath of hair follicles, and sweat glands. It localizes to the lateral and apical surfaces of cells.
As is typical of X-linked recessive disorders, expression is full blown in affected males, and carrier females may express none, some, or all of the features of the disorder, often in a patchy distribution. Between 60% and 80% of carrier females express some clinical signs of the disorder; the most frequent are patchy hypotrichosis and hypodontia. The disorder can be inherited from a carrier mother or occur in an affected individual as the result of a de novo mutation. Approximately 70% of affected males inherited the mutation from a carrier mother.
Mutations in an autosomal gene, EDAR, mapped to 2q11-q13, have been implicated in an autosomal dominant form of hypohidrotic ectodermal dysplasia (HED; OMIM #129490) and in an autosomal recessive form (OMIM #224900)10 that are clinically similar to XLHED. Both these entities are much rarer. EDAR acts as a receptor for ectodysplasin. Mutations in yet another gene, EDARADD, have been identified recently in autosomal recessive HED and autosomal dominant HED.11,12 EDARADD is an intracellular adaptor protein that assists in transmitting the signal from the activated EDA receptor to the nucleus of the cell. Individuals with autosomal dominant HED appear to have a milder defect in the ability to sweat.
Certain mutations in the X-linked NEMO gene, which causes incontinentia pigmenti (IP) in females, have been shown to result in HED and immune defects in males (see Chapter 143).13,14
Affected males may present at birth with a collodion membrane or with marked scaling of the skin,15 similar to congenital ichthyosis. Scalp hair is usually sparse, fine, and blonde. It may thicken and darken at puberty, and secondary sexual hair is typically normal. Other body hair is usually sparse or absent.
The ability to sweat is significantly compromised, and most affected males have marked heat intolerance. Sweat pores are usually undetectable on physical examination, and fingerprint ridges are effaced. The inability to sweat adequately in response to environmental heat results in an elevation of core temperature and bouts of unexplained high fevers, usually leading to an extensive workup for infectious disease, malignancy, or autoimmune disease before the correct diagnosis is recognized. In an older series of patients, mental retardation was reported as a feature of XLHED. Currently, this is believed to have been due to damage from prolonged high fevers and seizures and not to be an intrinsic feature of the disorder.16
The nails are usually normal; reports of thin, fragile nails are not convincing.17 Periorbital wrinkling and hyperpigmentation are typical and often present at birth (Figs. 142-2A–142-2C). Eczema affects more than two-thirds of affected males and is often difficult to manage. Hyperplasia of sebaceous glands, particularly on the face, can develop over time and appear as small, pearly, flesh-colored to white papules that may resemble milia.
Figure 142-2
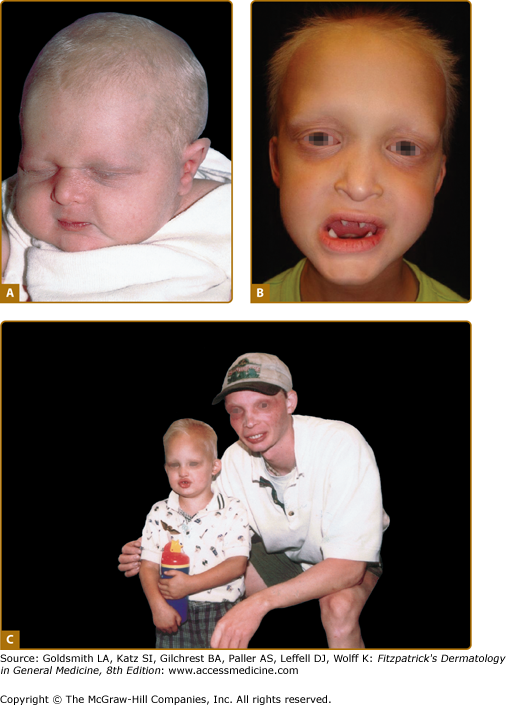
Hypohidrotic ectodermal dysplasia. A. Newborn with periorbital wrinkling, beaked nose. Diagnosis would not be suspected unless there were a positive family history. B. Peg-shaped teeth; fine periorbital wrinkling can be appreciated. C. Two unrelated males with X-linked hypohidrotic ectodermal dysplasia; adult is wearing dentures; periorbital wrinkling and hyperpigmentation are evident. (Used with permission from the National Foundation for Ectodermal Dysplasias.)
Hypodontia, oligodontia, or anodontia are invariable features of XLHED in affected males. Hypoplastic gum ridges in an affected infant can be an early clue to the diagnosis of the disorder. Teeth that do erupt are usually peg-shaped and small (see Fig. 142-2B). The facies of the disorder are characterized by frontal bossing and a depressed midface with a saddle nose and full, everted lips. Otolaryngologic manifestations include thick nasal secretions and impaction, ozena, sinusitis, recurrent upper respiratory tract infections and pneumonias, decreased saliva production, hoarse voice, and an increased frequency of asthma. The increased frequency of respiratory tract infections has been attributed to hypoplastic or absent mucus secreting glands in the bronchial tree.18 Gastroesophageal reflux and feeding difficulties may be a problem in infancy. The basis for this is unknown. Preliminary studies suggest that there may be failure to thrive in infancy and early childhood in as many as 20% to 40% of affected boys, with catch-up growth seen later.19 Although several reviews have suggested that infant mortality may be increased, unbiased confirmatory data are lacking.
Female carriers for XLHED may be affected as severely as males or show few, if any, signs of the disorder (Fig. 142-3). Heat intolerance, if present, is usually mild; adult carrier women comment that they do not sweat much or that they do not like very warm weather, but it is unusual for a female to experience fever due to inability to sweat. Typically, a few teeth may be peg-shaped or missing and scalp hair may be patchy or thin. Careful examination of the skin of carrier females often reveals a diminution in or patchy distribution of sweat pores. This sometimes can be appreciated readily just by magnification of fingertip pads or may require more sophisticated sweat testing.
Figure 142-3
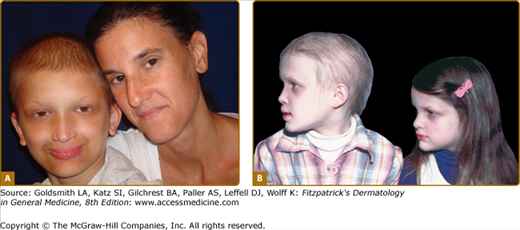
Hypohidrotic ectodermal dysplasia. A. Female carrier of X-linked hypohidrotic ectodermal dysplasia with her affected son. B. Two sisters with X-linked hypohidrotic ectodermal dysplasia manifesting to different degrees. Note periorbital hyperpigmentation, full everted lips, and sculpted noses. (From Sybert VP Genetic Skin Disorders. New York, Oxford University Press, 1997.)
As noted in Section “Etiology, Pathogenesis, and Genetics,” the autosomal dominant and autosomal recessive forms of HED are similar, although the autosomal dominant form may be milder. The X-linked form is by far the most common and always should be the diagnosis of default in a sporadic case.
The epidermis is thinned and flattened. There is a reduction in the number of sebaceous glands and hair follicles. Eccrine glands are absent or incompletely developed. Histologic evaluation of the skin is usually not necessary.
|
The scaling skin at birth may result in a misdiagnosis of congenital ichthyosis. Repeated bouts of fever may be thought to have an infectious source. The diagnosis of HED is recognized readily when expected, such as when an at-risk male is born into a family in which the disorder is known to segregate. Examination for sweat pores and a Panorex view of the jaw leads quickly to the correct diagnosis. Absence of eccrine structures in skin biopsies of the scalp and/or palmar region may help confirm a diagnosis,20 but are rarely necessary. In an isolated, fully expressing female, the autosomal dominant and recessive forms of HED need to be considered. Family history is mandatory, and mothers always should be examined fully to detect mild manifestations of the X-linked form. Molecular testing may be helpful but is not indicated for clinical diagnosis in most instances. Referral to a pediatric dermatologist may be helpful if the diagnosis is in question.
Maintenance of cool ambient temperatures is vital to prevent hyperpyrexia. Most children do well with simple measures, such as wet T-shirts, air conditioning in home and school, wet headbands, etc. Occasionally, cooling vests allow a broader range of participation in sports and vigorous physical activity in warm climates.
Dental restoration is of primary importance, and early implementation of dentures and ultimate use of dental implants are mainstays of treatment.
Management of otolaryngologic complications, asthma, and recurrent infections needs to be individualized. The eczema may be quite refractory to care.
The National Foundation for Ectodermal Dysplasias (http://www.nfed.org) has several pamphlets for individuals and professionals dealing with diagnosis and treatment. |
Although infancy and childhood are complicated by many problems, most individuals with HED lead adult lives that allow them to function successfully in society. Heat intolerance seems to decrease due to the development of some ability to sweat in adolescence or to the development of common sense and adaptation of lifestyle, or both.
Recombinant ectodysplasin protein injections of pregnant affected (Tabby) mice led to permanent rescue of the phenotypic features in the offspring.21 Further, early postnatal injections were also shown to ameliorate the developmental defects. Similar results have been seen in affected canine models who were infused with recombinant ectodysplasin protein in the postnatal period, with resultant normalization of teeth, lacrimation and sweating ability, as well as decreased eye and respiratory infections.22,23 These developments in animal models have been encouraging and human trials are planned. An international patient registry has been established by the NFED to facilitate these clinical trials.
Hidrotic Ectodermal Dysplasia (Clouston Syndrome; OMIM #129500)
Hidrotic ED was first described in a French–Canadian kindred.24 It has been reported in other ethnic groups, but the majority of affected individuals can trace their ancestry back to an original French–Canadian settler.
Hidrotic ED is autosomal dominant with variable expression (the degree of severity can vary within and between families). Males and females are affected in equal numbers and to equal degree. The gene maps to the centromeric region of the long arm of chromosome 13.
The disorder is caused by mutations in a connexin gene, GJB6 or connexin-30.25 Different mutations in the same gene are responsible for a form of nonsyndromic autosomal dominant deafness and at least one patient with keratitis-ichthyosis-deafness (KID syndrome) (see Chapter 49). Other connexin genes show similar variability in mutation: disease correlations [e.g., mutations in connexin-31 (GJB3)] can cause either erythrokeratodermia variabilis (see Chapter 49) or late-onset autosomal deafness.
The scalp hair is wiry, brittle, and pale, and there is often patchy alopecia (Fig. 142-4A). This progresses in adult life and may lead to total alopecia. Body and facial hair are affected. The nails may be milky white in infancy and early childhood, gradually thickening and becoming dystrophic. The nail plates in adults are thick, short, and slow growing. They separate distally from the nail bed (see Fig. 142-4C), and may cause pain. Anonychia has been reported. Not all the nails are necessarily affected to the same degree. Progressive palmar/plantar hyperkeratosis is common (see Fig. 142-4B). In contrast to HED, sweating is normal, as are the teeth. Oral leukoplakia has been reported. Conjunctivitis and blepharitis, possibly due to poor function of sparse eyelashes, are common.
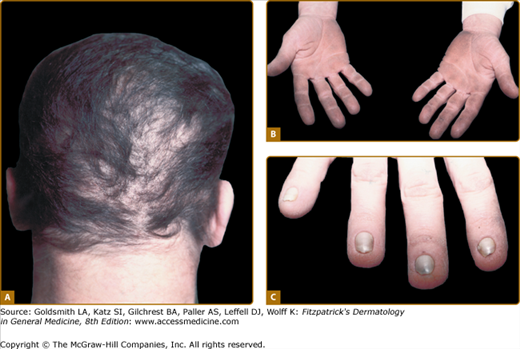
The thickened palms and soles show orthohyperkeratosis with a normal granular layer. On electron microscopy, an increase in the number of desmosomes in the cells of the stratum corneum is found. The hair shows nonspecific changes.
(See Box 142-1)
The diagnosis is straightforward. The involvement of nails and hair and palmar/plantar thickening, in the absence of other signs of ED, are reasonably specific. Other palmar/plantar hyperkeratoses do not have similar hair changes. Orofacial clefting differentiates other forms of autosomal dominant hidrotic ED, such as ankyloblepharon–ED–cleft palate (AEC) syndrome or Rapp–Hodgkin syndrome. Although the nail changes are similar to those of pachyonychia congenita, the hair changes are distinctive.








