Abstract
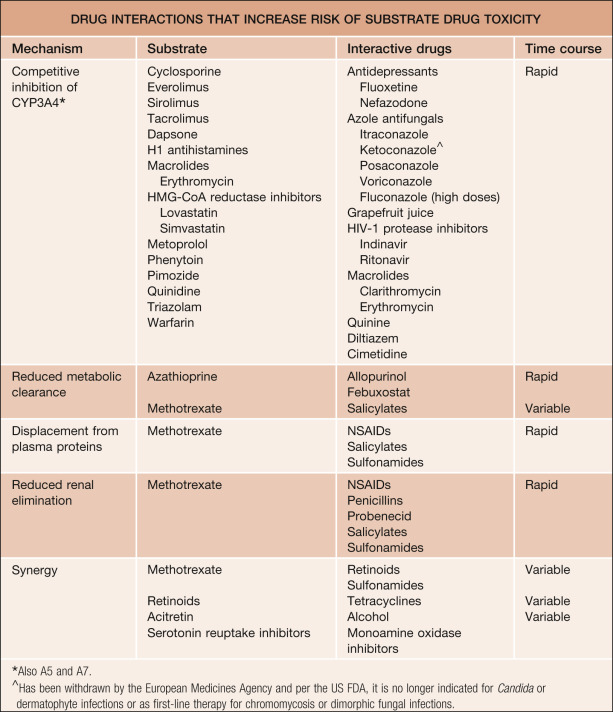
Interactions between or amongst multiple therapies may lead to increased toxicity, decreased efficacy, or both. Knowledge of the interactive properties of drugs can help prevent serious adverse drug interactions. Non-prescription drugs, herbal or alternative medicines, and foods (e.g. grapefruit juice) may also be implicated in drug interactions. Prediction of drug interactions is possible when those agents likely to produce alterations in drug metabolism via inhibition or induction of the cytochrome P450 (CYP) system are recognized. The CYP enzymes are the most important drug-metabolizing enzymes. They are present in the endoplasmic reticulum of many types of cells, but are at highest concentration in hepatocytes. Fortunately, the metabolism of most drugs can be accounted for by a relatively small subset of the CYP isoforms. It is thought that over 90% of human drug oxidation can be attributed to six enzymes (isoforms): CYP1A2, 2C9/10, 2C19, 2D6, 2E1, and 3A4. One-third to one-half of drug metabolism can be attributed to CYP3A4. Knowledge of the substrates, inhibitors, and inducers of these enzymes assists in predicting clinically significant drug interactions. The isoform CYP2D6 is involved in about one-fourth of all drug metabolism. The concept that most drug oxidation reactions are catalyzed primarily by a small number of CYP enzymes is important in that approaches to identifying drug–drug interactions become more feasible, both in vitro and in vivo .
Knowledge of the interactive potential of various drugs can help prevent serious adverse drug interactions. Drug interactions primarily involve an “object drug” (“bullet”, substrate) that is the drug affected by the interaction and “precipitant drug” (“accomplice”, inhibitor, inducer), which is the drug causing the interaction. The most important “precipitant” drugs interfere with drug absorption, distribution, metabolism, and elimination. Foods, such as grapefruit juice, are also important precipitants. Many of these drug combinations can be administered safely with appropriate dosage adjustments or by substitution with another member of the drug class with less potential for drug–drug interactions.
The clinical importance of specific drug interactions is often either overestimated or underestimated, as these assessments are largely based on clinical experience in using the particular drug combination. The clinical outcome of most drug interactions is highly situational, and most patients who receive drugs with the potential for interactions do not develop adverse effects. Emphasis should be placed on those factors that increase or decrease the risk for a given patient.
While it is clearly not possible for individual clinicians to remember all drug interactions, it is important to understand how to use the drug interaction information provided by electronic medical record programs, PubMed, Micromedex ® Drug Reference, books, and other sources.
Keywords
drug interactions, drug absorption, drug distribution, drug metabolism, cytochrome P450, CYP3A4, P-glycoprotein, cytochrome P450 inhibition, cytochrome P450 induction, genetic polymorphisms
Introduction
There are two concerns in drug safety: drug reactions and drug interactions. When multiple medications are prescribed, drug interactions become an important safety and efficacy consideration for patients and physicians alike . Non-prescription drugs, as well as herbal or alternative medicines and foods (such as grapefruit juice), may also be implicated in interactions with drug therapy.
It is difficult to obtain precise rates of incidence and prevalence, as specific diagnostic codes for drug interactions are lacking . Drug interactions are known to be responsible for up to 2.8% of hospital admissions . It has been estimated that adverse drug outcomes occur about once every 100 patient days . Although it is impossible to remember all potential drug interactions, knowledge of the interactive properties of drugs can help reduce the risk of serious adverse outcomes. Moreover, it is the responsibility of physicians to counsel patients regarding drug interactions . Unfortunately, even when serious new drug interactions are recognized and reported, physicians, pharmacists and patients are often unaware that there is a risk .
Putting Interactions Into Perspective
Prescribing drugs with the potential for a deleterious interaction increases the risk of, but need not lead to, an adverse outcome. Many drug–drug interactions are susceptible to control by dose adjustment. Some interactions can even be exploited for a therapeutic advantage, e.g. coadministration of erythromycin to increase cyclosporine levels as a means of reducing costs associated with the latter.
While most drugs are associated with interactions, many do not produce significant outcomes . In fact, not all listed or reported drug interactions are actually clinically significant. Whereas some have little clinical relevance, others are clearly defined as contraindications on the basis of substantiation of risk, potential severity, and frequency of occurrence. Still others can be successfully managed by dosage adjustment and monitoring. In one study that examined the occurrence of drug interactions, based upon adverse events reported during large clinical trials , the authors concluded that serious adverse drug reactions (ADRs) secondary to drug–drug interactions were infrequent; however, drug–drug interactions that involved selected drugs with a narrow therapeutic index (which elicited life-threatening undesired effects) did occur. Drug–drug interactions are the most common cause of medication errors in high-income countries, with a prevalence of 20–40%, especially due to polypharmacy in the elderly . Thus, for most drug interactions, it is necessary to assess each patient situation individually.
Assessment of Risk in the Clinical Outcome of Drug Interactions
The clinical importance of specific drug interactions is often either over- or underestimated, as these assessments are largely based on clinical experience in using a particular drug combination . The clinical outcome of most drug interactions is highly situational, as most patients who receive drugs with the potential for interactions do not develop adverse effects. Emphasis should be placed on those factors that increase or decrease the risk to a given patient.
In order to prevent or detect drug interactions, the physician needs to identify risk factors in each individual patient. That said, some patient groups are more likely than others to develop adverse events caused by drug interactions. Identified risk factors are listed in Table 131.1 and are grouped by category .
| PATIENT RISK FACTORS FOR DRUG INTERACTIONS |
|
The elderly frequently experience drug interactions because of the physiologic changes that accompany the aging process and the types of drugs that older patients tend to receive . Polypharmacy, which is common in the elderly, makes them particularly susceptible. In addition, medications may impair pathways of drug elimination by interfering with drug metabolism, thereby increasing the likelihood of adverse drug reactions. Alterations due to advanced age, including changes in drug–protein binding and drug distribution within tissue, may also promote drug interactions. Of note, most adverse metabolic drug interactions occur when an inhibitor or inducer is begun in a patient who previously had stable levels of a substrate drug.
HIV-infected patients also have a high rate of adverse drug reactions . In some instances, this may relate to phenotypic changes in drug metabolism, which can vary with progression of the underlying HIV infection . The latter can alter enzyme function, with resultant changes in drug metabolism and a higher rate of adverse reactions in these patients.
A major source of interindividual differences in drug metabolism is genetic polymorphisms, which are inherited and can result in significant variations in the activity of drug-metabolizing enzymes. For example, the genes encoding cytochrome P450 (CYP) isoforms are highly polymorphic (see Table 131.13 ). In some cases, it is actually possible to determine an individual’s genotype . Further details on genetic polymorphisms are discussed later in the chapter.
Interactions with other drugs can predispose a patient to the development of certain types of ADR. For example, coadministration of valproic acid increases the risk of severe cutaneous adverse reactions to lamotrigine , and allopurinol increases the risk of exanthematous eruptions to antibiotics such as amoxicillin. A disease state itself may directly affect the likelihood of an ADR. For example, active infection with Epstein–Barr virus or cytomegalovirus also increases the risk of exanthematous eruptions to amoxicillin. The basis of these interactions is unknown but may represent a combination of factors, including alterations in drug metabolism, drug detoxification, antioxidant defenses, and immune reactivity . The disease state may also dictate the way in which a drug is prescribed, and this will subsequently affect the outcome. When a drug has more than one therapeutic action, an interacting drug may affect the action of the first drug when it is used to treat one disease but not when it is used to treat the first disease. This is known as pharmacologic selectivity.
An example of intrinsic effects of disease states would be when epinephrine (adrenaline) is given to patients receiving non-cardioselective β-adrenergic blockers (but who do not have anaphylaxis) and this results in hypertension. In contrast, the same β-blockers inhibit the pressor response to epinephrine when the latter is given to patients with anaphylaxis .
An example of a dose-dependent drug interaction is the concomitant use of nonsteroidal anti-inflammatory drugs (NSAIDs) and methotrexate. Available evidence indicates that the risk of this combination is considerably greater in patients receiving high-dose methotrexate as cancer chemotherapy than it is in patients receiving lower weekly dosages for psoriasis . This is because the distribution of the drug is not very important mechanistically in determining drug interactions, but changes in drug elimination are. When larger doses of methotrexate (as used in cancer therapy) are given in combination with an NSAID, reduced drug elimination via the kidneys becomes an important issue, increasing the risk of methotrexate toxicity.
Gender-related differences in pharmacokinetics may cause variations in drug absorption, gastric emptying, and distribution based on percentage of adipose tissue . Gender-related differences in receptor density and sensitivity, enzyme activity (CYP2D6), and underlying disease activities also contribute to pharmacokinetic variation. The effect of obesity on metabolism is cytochrome-specific. For example, obesity decreases the activity of CYP3A4 and increases the activity of CYP2E1.
Certain medications are clearly more likely to be involved in drug interactions. More specifically, clinically significant interactions occur more frequently with drugs that have a narrow margin of safety, i.e. a narrow therapeutic window. Drugs with the potential for such serious interactions include warfarin and cyclosporine. When prescribing a new medication that could potentially interact with warfarin, it is prudent to have the patient’s international normalized ratio (INR) measured within 2–3 days of administration of the new drug.
Medication-related factors that contribute to clinical risk include the dose, route of administration and duration of administration of the precipitant drug (the drug that causes the interaction) as well as the sequence of administration of the interacting drugs. Most metabolic drug interactions are dose-related. That is, as the dose of the precipitant drug is increased, the magnitude of its effect on the object drug tends to increase. Thus, the dose of the precipitant drug is often an important determinant of risk. However, the dose of the object drug may also affect the risk of an adverse drug interaction. For example, a patient who takes small doses of an object drug with serum concentrations at the lower end of the target range is at a lesser risk when an enzyme-inhibiting precipitant drug is added, than would be a patient taking large doses of the same object drug. Lastly the bioavailability of either drug may have an impact. The route of administration is an important risk factor for some non-metabolic drug interactions, such as when one drug binds to another in the gastrointestinal (GI) tract. However, the route of administration can also be important for metabolic drug interactions, especially when the drug undergoes extensive first-pass metabolism in the gut wall and liver by CYP3A4 or P-glycoprotein (see below). Most drug interactions also have a typical time course over which the effects develop. For example, giving rifampin, a typical inducer of CYP3A4, for only a few days is unlikely to have much effect on substrates of CYP3A4, as induction of an enzyme takes weeks to occur.
In summary, there is pharmacodynamic and pharmacokinetic variability between people, as well as host variability in terms of disease state. Overall, this variability contributes to confusion, as tables and lists that outline potentially interacting medications vary amongst different sources. One reason is that various levels of evidence exist for many of these drugs in terms of their ability to contribute to/cause drug interactions. This is outlined in more detail in the next section.
Levels of Evidence
Drug interaction literature is often confusing due to poorly substantiated claims . This confusion occurs as a result of inaccurate or cursory evaluations of published cases or inappropriate extrapolations from the literature. Metabolic drug interactions are a major source of potential clinical problems, but their investigation during drug development is often incomplete. In vitro studies give very accurate data on the interactions of drugs with selective CYP isozymes, but their interpretation in the clinical context is difficult. Although in vitro systems have been developed to test the effects of certain drugs on the metabolism of other drugs, these systems may not accurately predict the effect in patients receiving drugs with complex metabolism. Also, problems with the detection of adverse events after a drug has been released arise mainly because such events are rare. It takes a surveillance system with a high degree of sensitivity to detect such problems .
Furthermore, most in vivo and in vitro studies of drug interactions evaluate two-drug regimens, and the results may not apply to the multidrug regimens used clinically. This is especially true for a regimen consisting of three or more drugs with opposing effects on CYP3A4 metabolism. The lack of studies of multiple drug interactions provides little assistance to the prescribing physician, who is left to rely on adverse events or treatment failure to demonstrate whether an interaction has occurred. In addition, the design of in vivo studies is sometimes poor (choice of prototype substrate, doses, schedule of administration, number of volunteers), with the risk of minimizing the real potential for interaction. To link in vitro and in vivo studies, several authors have suggested using extrapolation techniques, based on the comparison of in vitro inhibition data with the active in vivo concentrations of the inhibitor. However, the lack of knowledge with regard to one or several important parameters, such as the role of metabolites and intrahepatocyte accumulation, often limits the ability to make safe and accurate predictions.
The uncertainty and inaccuracy of predicting the extent and duration of in vivo drug interactions currently stems from a lack of definitive models by which to assess likely substrate and inhibitor concentrations at the active site of metabolism. Additional issues contributing to the uncertainty of predicting drug interactions include assumptions of the contribution of presystemic drug extraction and the effect of inhibitors on the processes involved. As a consequence, these methods are useful for complementing in vivo studies and helping to design clinically relevant in vivo studies, but in the foreseeable future they will not totally replace in vivo investigations.
Absorption
Interactions that alter the absorption of drugs often lead to dramatic changes in plasma drug concentrations. Drug interactions within the GI tract can result in decreased absorption. This reduces the bioavailability or the amount of drug available to the systemic circulation and results in subtherapeutic serum concentrations. The underlying mechanisms of most drug interactions that alter GI absorption involve: (1) the formation of drug complexes that reduce absorption; (2) alterations in gastric pH; and/or (3) changes in GI motility that alter transit time .
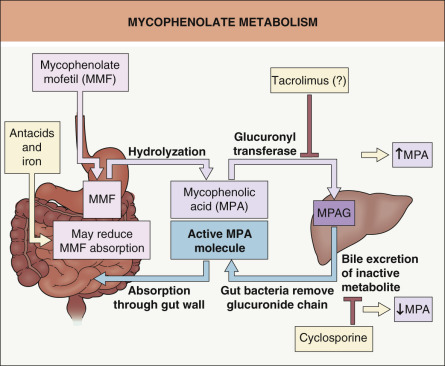
Common drugs that form complexes with other drugs include antacids, sucralfate, and bile acid sequestrants (e.g. cholestyramine, colestipol, colesevelam) ( Table 131.2 ). A significant interaction occurs between multivalent cations – such as calcium, aluminum, iron and magnesium – and tetracyclines or fluoroquinolone antibiotics. For example, there is an 85% reduction in the absorption of ciprofloxacin when ingested 5–10 minutes after a dose of an aluminum hydroxide/magnesium hydroxide antacid . These interactions can be easily avoided by administering the fluoroquinolone at least 2 hours before or 6 hours after the antacid or iron. Alendronate, as well as other bisphosphonates prescribed for the prevention and treatment of osteoporosis, forms complexes with cations and several other drugs, thereby further decreasing their already low oral absorption. Once-weekly dosing reduces the opportunity for bisphosphonate-related interactions. When mycophenolate mofetil and iron preparations were administered concomitantly, a significant decrease in mycophenolate mofetil absorption was observed ( Fig. 131.1 ) .
| DRUG INTERACTIONS THAT REDUCE THE EFFICACY OF SUBSTRATES | |||
|---|---|---|---|
| Mechanism | Substrate/parent drug | Concomitant drugs | Time course |
| Reduced GI absorption | Itraconazole Ketoconazole ^ | Antacids * Didanosine H 2 antihistamines Proton pump inhibitors Sucralfate | Rapid |
| Fluoroquinolones | Antacids * Iron Sucralfate | ||
| Tetracycline Bisphosphonates | Divalent cations | ||
| Calcium | |||
| Magnesium | |||
| Iron | |||
| Dapsone | Didanosine | ||
| Mycophenolate mofetil | Iron Antacids * | ||
| Induction of CYP3A4 † | Calcineurin inhibitors Cyclosporine Tacrolimus Oral contraceptives Corticosteroids Dexamethasone Methylprednisolone PrednisoneWarfarin | Anticonvulsants Carbamazepine Phenytoin Phenobarbital Antituberculous agents IsoniazidRifampin Dexamethasone Griseofulvin | 1–2 weeks |
| Antagonistic effects | Epinephrine | β-Blockers | |
| Cyproheptadine |
| ||
^ Has been withdrawn by the European Medicines Agency and per the US FDA, it is no longer indicated for Candida or dermatophyte infections or as first-line therapy for chromomycosis or dimorphic fungal infections.
* Often contain aluminum hydroxide, magnesium hydroxide and/or calcium carbonate.
Drugs that increase gastric pH, such as proton pump inhibitors, antacids and H 2 antihistamines, may reduce the absorption of drugs such as itraconazole, posaconazole and ketoconazole, which are best absorbed in an acidic environment . Although itraconazole is best absorbed when the gastric pH is low, its administration with food is more important for achieving high plasma concentrations . Of note, the absorption of fluconazole is unaffected by variations in gastric pH. Similarly, the coadministration of drugs that can increase gastric pH (see above) with atazanavir and raltegravir are not recommended. Drugs that affect GI motility, such as anticholinergic agents, may decrease the rate of absorption but not the extent of absorption. An overall reduction in drug absorption has more clinical significance .
Some drugs may interfere with the enterohepatic recirculation of a substrate drug. When the substrate is excreted into the GI tract, a second drug can bind to it and prevent its reabsorption back into the systemic circulation. The bound substrate drug is excreted in the feces, thereby effectively shortening its half-life. An example of this is the concurrent administration of warfarin and bile acid sequestrants (e.g. cholestyramine, colestipol, colesevelam) in which the half-life of warfarin is shortened.
P-glycoprotein (PGP)
Membrane-bound transport systems may also determine drug disposition . These transporters are found in multiple tissues and actively pump drug molecules either out of cells (efflux) or into cells (uptake). PGP is an ATP-dependent plasma membrane glycoprotein belonging to the superfamily of ATP-binding cassette transporters that functions primarily as an efflux pump ( Fig. 131.2 ) . Other transporters include the organic anion and organic cation transporters. In humans, the multidrug resistance (MDR) genes, including MDR1 , encode membrane glycoproteins that function as drug transporters and hence affect both drug absorption and elimination.
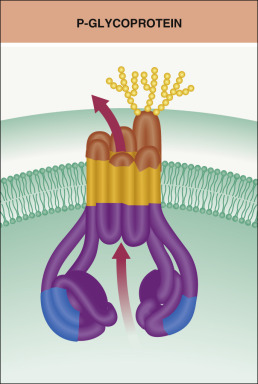
High levels of PGP are found in superficial columnar epithelial cells of the small intestine, the apical surface of epithelial cells in the proximal tubules of the kidney, and in the biliary canalicular membrane of hepatocytes. PGP is also detected in high concentrations in the endothelial cells of the capillaries of the blood–brain barrier, testes, uterus and placenta. An understanding of the physiologic regulation of these transporter proteins is key to designing strategies for improving the therapeutic efficacy of drugs that serve as their substrates ( Table 131.3 ).
| P-GLYCOPROTEIN SUBSTRATES | ||
|---|---|---|
| Antimicrobials | HIV protease inhibitors | Cardiac agents |
| Ciprofloxacin | Indinavir | Amiodarone |
| Erythromycin | Nelfinavir | Atorvastatin |
| Ivermectin | Ritonavir | Digoxin |
| Other quinolones | Saquinavir | Diltiazem |
| Rifampin | Antiemetics | Lovastatin |
| Anticancer agents | Domperidone | Nadolol |
| Actinomycin D | Ondansetron | Pravastatin |
| Daunorubicin | Rheumatologic agents | Propranolol |
| Docetaxel | Colchicine | Quinidine |
| Doxorubicin | Methotrexate | Timolol |
| Etoposide | Quinine | Verapamil |
| Mitomycin C | Immunosuppressives | Miscellaneous |
| Paclitaxel | Cyclosporine | Cimetidine |
| Vinblastine | Tacrolimus | Lidocaine |
| Vincristine | Loperamide | |
| Terfenadine * | ||
These membrane-bound transport systems appear to have developed as a mechanism for protecting the body from harmful substances. It appears that PGP acts as a pump whereby the efflux of drugs from the cell membrane or cytoplasm is powered by the energy from ATP hydrolysis. For example, the aminoglycoside antibiotics amikacin and tobramycin are not effectively delivered orally, perhaps because of active efflux from the brush border cells of the small intestine by the PGP pump. The most remarkable property of PGP is its ability to transport a diverse array of compounds that do not appear to share obvious structural characteristics. The range of substrates, inhibitors, and inducers of PGP is vast and expanding ( Tables 131.3 & 131.4 ).
| INHIBITORS OF P-GLYCOPROTEIN | ||
|---|---|---|
| Antimicrobials | Psychotropic agents | Cardiac agents |
| Clarithromycin | Amitriptyline | Amiodarone |
| Erythromycin | Chlorpromazine | Carvedilol |
| Itraconazole | Desipramine | Diltiazem |
| Ivermectin | Disulfiram | Dipyridamole |
| Ketoconazole | Doxepin | Felodipine |
| Mefloquine | Fluphenazine | Nicardipine |
| Ofloxacin | Haloperidol | Nifedipine |
| Posaconazole | Imipramine | Propranolol |
| Rifampin | Steroid hormones | Verapamil |
| Voriconazole | Progesterone | Miscellaneous |
| Immunosuppressives | Testosterone | Grapefruit juice |
| Cyclosporine Tacrolimus | Orange juice isoflavones | |
| Ritonavir | ||
| Tamoxifen | ||
Because PGP blocks absorption in the gut, these glycoproteins should be considered part of the “first-pass effect”. In fact, PGP can “set up” or act as “gatekeepers” for later cytochrome P450 actions. Although the inhibition and induction of intestinal CYP3A enzymes from metabolic processes result in direct changes in drug absorption, the inhibition and induction of PGP primarily affects the rate of drug absorption . If one drug is a substrate of both PGP and CYP3A4 (which are found in close proximity in the intestinal wall), and a second drug is added that is an inhibitor of both PGP and CYP3A4 (e.g. erythromycin, ketoconazole), then a greater amount of the first drug will be absorbed. Because CYP3A4 is inhibited, higher levels of unmetabolized drug will enter the blood. The effect of PGP blockade is to “open the gates” so that the later actions of CYP3A4 inhibition will be increased.
PGP is an important component of the blood–brain barrier and an active PGP will prevent drugs from entering the brain. It has been suggested that the reason newer antihistamines do not cause sedation is that PGP activity acts as a barrier to CNS penetration. This would suggest that PGP inhibitors (see Table 131.4 ) could interact with and allow increased cerebral concentrations of these antihistamines, with an attendant increase in sedation .
Evidence also suggests that intestinal PGP plays a significant role in the first-pass elimination of cyclosporine, probably by being a rate-limiting step in absorption. Intestinal CYP3A4 is thought to play a lesser role . However, the overlap of tissue distribution and substrate specificity of CYP3A4 and PGP in the intestinal wall makes it difficult to define the precise mechanisms of some drug interactions and to predict the plasma concentrations of certain drug combinations. Moreover, the involvement of CYP3A4 and PGP in drug interactions is not always complementary.
Distribution
Drugs that are highly protein bound (>90%) may cause interactions based on alterations in distribution. When one drug displaces another from plasma protein-binding sites, the free serum concentration of the displaced drug is increased and its pharmacologic effect increases. However, the unbound fraction of the drug is not only more available to sites of action but is also more readily eliminated. Any enhanced pharmacologic effect occurs only transiently because of a compensatory increase in elimination, and the effect of displacement interactions is then negligible . Therefore, interactions involving drug displacement from binding proteins tend to be self-limiting . Typically, the pharmacologic activity of the displaced drug is increased for a few days. This is followed by a return of the pharmacologic response back to the previous unbound serum concentration, even if the concomitant therapy is continued. Therefore, it is safe to say that if a patient does not manifest an adverse event from the combination therapy in the first week or so of administration, an adverse event probably will not occur.
In practice, protein-binding displacement interactions do not produce clinically important changes in drug response unless the drug also has a limited distribution in the body, is slowly eliminated, or has a low therapeutic index . For this reason, protein-binding displacement interactions may assume greater importance when the displacing drug also reduces the elimination of the substrate drug. Good examples of this principle are the interactions between NSAIDs and methotrexate ( Table 131.5 ).
Medications that are most susceptible to interactions based on changes in drug distribution involving displacement from binding proteins include warfarin, sulfonamides and phenytoin .
Drug Biotransformation
Cytochrome P450 Enzymes
After their administration, drugs are metabolized through a series of reactions to enhance their hydrophilicity and to facilitate excretion. These drug biotransformation reactions are broadly grouped into two phases, I and II. Phase I reactions involve intramolecular changes such as oxidation, reduction and hydrolysis that make the drug more polar and therefore more readily eliminated. Phase II reactions are conjugation reactions in which an endogenous substance combines with the functional group derived from phase I reactions to produce a highly polar drug conjugate that can be even more readily eliminated. These reactions involve glucuronidation and sulfation.
The cytochrome P450 (CYP) enzymes are the major drug-metabolizing enzymes ( Fig. 131.3 ). They are present in the endoplasmic reticulum of many cells but their highest concentrations are found in hepatocytes . CYP enzymes are also present in the crypt cells of the GI tract, with the highest concentrations found in the enterocytes at the tips of the villi; their presence accounts for the first-pass metabolism of many drugs. These heme-containing proteins are encoded by a gene superfamily, with the encoded isoforms exhibiting distinct but overlapping substrate specificities and isoform-specific regulatory and pharmacogenetic properties . The nomenclature employs a three-tier classification consisting of the family (40% homology in amino acid sequence), the subfamily (~75% homology), and the individual protein (e.g. CYP2D6).
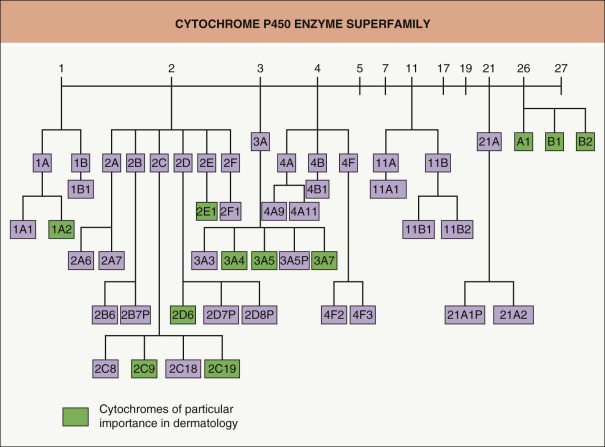
An increased understanding of CYP drug metabolism has solved much of the mystery behind drug interactions. While there are approximately 60 genes that encode CYP isoforms, over 90% of drug oxidation can be attributed to six main cytochromes: CYP1A2, 2C9, 2C19, 2D6, 2E1 and 3A4 . The metabolism of a drug by a specific isoenzyme indicates that it is a substrate for that enzyme. Whether enzyme inhibition or induction occurs is an entirely separate issue . Many drugs serve only as substrates and produce no significant enzyme inhibition or induction. It is entirely possible for a drug to be a substrate for one enzyme and inhibit or induce another enzyme that is not involved with its own metabolism. Therefore, drug interactions are more aptly termed drug–protein–drug (food) interactions. These are affected by: genetics (polymorphic genes cause particular enzymes to be less effective, 2D6 being an example); drugs (a drug may inhibit or induce a cytochrome, or interfere in the chemical pathway of another drug, e.g. itraconazole reduces cyclosporine metabolism by inhibiting CYP3A4); chemicals (dioxin is an inducer of CYP3A4 while a food such as grapefruit juice is an inhibitor of CYP3A4); and the environment (cigarette smoke is an inducer of CYP1A2). Deciding what is clinically relevant is a challenging, relatively new field of investigation.
Drug metabolism is investigated even before human exposure. With recombinant human CYP enzymes, it is possible to determine the metabolic pathways, potential genetic polymorphisms, ability to induce or inhibit drug metabolism, and possible drug interactions. Although there are limitations to the information gleaned from in vitro studies, nonetheless this information can be used to guide more expensive in vivo studies.
However, using in vitro tests that focus on cytochrome enzymes alone to predict clinical interactions may not always be reliable for a variety of reasons. First, it is not always possible to know the therapeutic concentration of a new drug and its primary metabolites in specific tissues . Second, there are a large number of pathways and interactions, and it is impossible to test them all even in an in vitro system. Third, the demonstration of an in vitro effect does not tell physicians whether that effect is likely to occur in clinical practice, i.e. the clinical significance of an in vitro interaction is unknown. Fourth, the underlying disease state may contribute to the development of a drug interaction and this would be unaccounted for by in vitro studies alone. Until clinical data demonstrate the presence or absence of a clinically significant interaction, dosage adjustments are premature .
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


