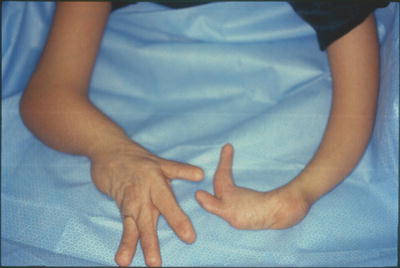
Fig. 12.1
A cross section of limb a bud showing the interactions of the two key players of the dorsal–ventral axis: EN-1 (ENGRAILED-1) and WNT7A
The ENGRAILED-1 Pathway and Dorsal Dimelia
Al-Qattan [2] defined the ENGRAILED-1 pathway (Fig. 12.2). In this pathway, EN-1 is a “transcription factor.” In other words, it is expressed following the action of a “ligand” on a “receptor.” The “ligands” are Bone Morphogenetic Proteins 4 and 7 (BMP4, 7) that act on the receptor BMPR1A. This results in the expression of EN-1 in the ventral ectoderm. EN-1 will have three main functions: induction of the AER, development of ventral structures of the hand, and restriction of WNT7A to the dorsal ectoderm. Disruption of this pathway will result in the lack of EN-1 functional expression in the ventral ectoderm. Ventral structures will not develop; and the unrestricted WNT7A will be expressed in the dorsal as well as the ventral ectoderm. Ectopic ventral WNT7A will induce the ectopic expression of LMX1B in the ventral mesoderm. The end result is “dorsal dimelia,” which is a hand with dorsal structures on the ventral aspect.
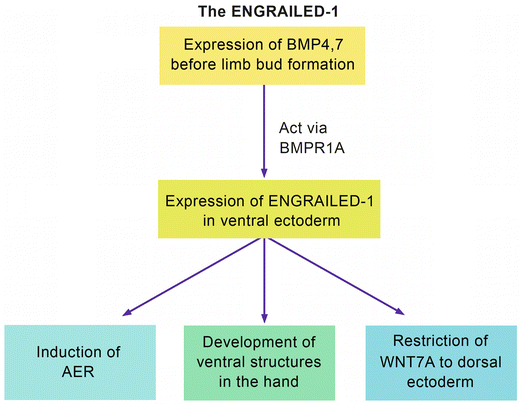
Fig. 12.2
The EN-1 pathway
Dorsal Dimelia in Experimental Animals
Experimental dorsal dimelia is induced by disruption of the EN-1 pathway. It is important to note that these animals will show dorsal dimelia of all digits in the fore- and hind-feet. In other words, each digit will show a double nail: one normal dorsal nail and another ectopic ventral nail. Experimental dorsal dimelia was induced in Bmpr1a conditional knockout animals [6], null mutations of En-1[7], mis-expression of Wnt7a in the ventral ectoderm [8], and mis-expression of Lmx1b in the ventral mesoderm [9].
Dorsal Dimelia in Humans
Dorsal dimelia in humans may be classified into two main groups: distal dorsal dimelia and proximal dorsal dimelia [10].
Proximal Dorsal Dimelia in Humans
Al-Qattan et al. [11] described one Egyptian family with isolated dorsalization of the skin of the proximal palm and the instep of the sole of the foot. Inheritance was autosomal dominant. Fingers and toes were completely normal. Hand function and gait were also normal. The proximal palm had a subcutaneous hamartoma with hyperpigmented hairy skin. Linkage analysis/exome sequencing showed an R584w variant in the GLE1 gene. GLE1 is involved in mRNA export; and RNA in situ hybridization showed a high Gle-1 expression in mouse embryo ventral cells and somites.
Distal Dorsal Dimelia in Humans
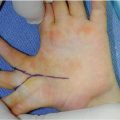
Distal dorsal dimelia is characterized by dorsalization of the distal palm and digits [12]. When fully expressed, the digit will have an ectopic palmar nail (the palmar and dorsal nails meet at the tip) (Fig. 12.3), the palmar skin of the digit and distal palm will show thin hyperpigmented skin, digital flexion is lacking, and the X-ray will show a tapering distal phalanx. The fully expressed phenotype is also known in the literature as “congenital palmar nail syndrome” [13, 14]. This fully expressed phenotype has amazing resemblance to the conjoined nail of Siamese “tripus” twins [15]. The twins have three lower limbs (hence the term tripus). Each twin has one normal lower limb on one side of the body. On the other side, the two adjacent lower limbs are fused into one. The conjoined feet will have eight separate toes, whereas the two big toes are fused into one digit. This digit will have all the features of “palmar nail syndrome.”
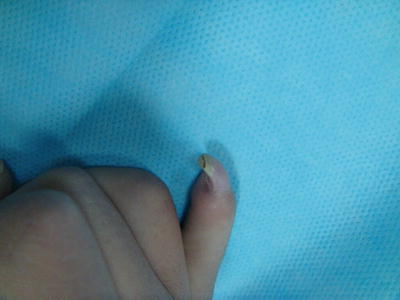
Fig. 12.3
Dorsal dimelia. Note the dorsal and ventral nails meeting at the tip of the little finger
Al-Qattan et al. [5] stressed that the clinical picture of distal dorsal dimelia in humans may not always be fully expressed. Hence, some cases may have isolated palmar nail while others may have isolated dorsalization of the palmar skin.
Al-Qattan and Kfoury [12] reviewed all cases of distal dorsal dimelia in humans and categorized them into three groups: syndromic, familial, and sporadic. Bilateral little finger dimelia has been described in syndromic patients with partial deletion the long arm of chromosome 6 [16] and Patau syndrome (trisomy 13) [17]. One case with DLX5 mutation and split-hand–split-foot malformation (also known as lobster-claw deformity) had dorsal dimelia of all digits [18]. The latter is the only human case with involvement of all digits. In all other human cases, dorsal dimelia involves either the ulnar or radial digits. Familial cases may have a family history of dorsal dimelia [19, 20] or ulnar ray deficiency [13, 21]. This is interesting because it links dorsal dimelia to SHH activity. In fact, several sporadic cases occurred in patients with ulnar ray deficiency [22, 23] or ulnar-sided cleft hand [5, 10, 24]. As expected, all these patients with ulnar defects showed dorsal dimelia in the ulnar digits.
However, several sporadic cases involving the ulnar digits occurred in patients with negative family history and no other concurrent anomalies [25–28]. Al-Qattan et al. [5] screened several sporadic cases with dorsal dimelia involving the ulnar digits for candidate genes such as loss-of-function mutations of BMP4, BMP7, BMPR1A, and EN-1, as well as gain-of-function mutations of WNT7A and LMX1B. However, the genetic analysis did not show any mutations. It was concluded that sporadic dorsal dimelia is probably a stochastic developmental error that is commonly seen with other concurrent hand malformations. In support of the latter statement, the author reported 3 cases of dorsal dimelia involving the radial digits; and in all cases there was radial-sided malformations such as thumb polydactyly [12], or radial ray deficiency [29, 30].
Management of Dorsal Dimelia
Proximal dorsal dimelia requires no treatment. In contrast, patients with distal dorsal dimelia may have cosmetic (the palmar nail) and functional (the lack of flexion) concerns. Options of management include conservative treatment (observation only) or surgery in the form of excision of the palmar nail along with pulp reconstruction or amputation of the distal phalanx [28]. Affected digits are usually held in extension with no active or passive flexion because of symphalangism. Osteotomy and fixation of the proximal interphalangeal joint in a more functional position is an option but no such procedure has been reported in the literature in patients with dorsal dimelia.
Dorsal dimelia of the index finger may occur in patients with absent thumb [29]. This poses a problem when there is need to pollicize the affected index finger. Anatomically, the affected finger has cartilaginous symphalangism of the interphalangeal joints. There is also a mirror-image flat extensor tendon on the palmar and dorsal aspects of the finger with no intrinsic muscle attachments. More important, there are two neurovascular bundles: one dorsal and one palmar. Al-Qattan and Kfoury reported these anatomical findings in a patient who had amputation of a duplicated digit with dorsal dimelia [12]; and these findings have obvious implications in the pollicization procedure.
The WNT7A Pathway and Ventral Dimelia
Figure 12.4 shows the WNT7A pathway. WNT7A (which is normally expressed in the dorsal ectoderm) acts at the cellular level by stimulation of specific receptors and the activation of two different pathways [2]: the calcium-mediated pathway and the beta-catenin (canonical) pathway. The former pathway leads to the expression of LMX1B in the dorsal mesoderm that will result in the normal development of dorsal structures in the hand. The latter pathway is responsible for maintaining SHH activity. As mentioned before, SHH is responsible for the development of the ulnar ray and the induction of FGF4 in the posterior part of the AER. The beta-catenin pathway also maintains the expression of another protein called ISLET 1. ISLET 1 is a major contributor to the initiation/outgrowth of the vertebrate hind limb and pelvis [31].
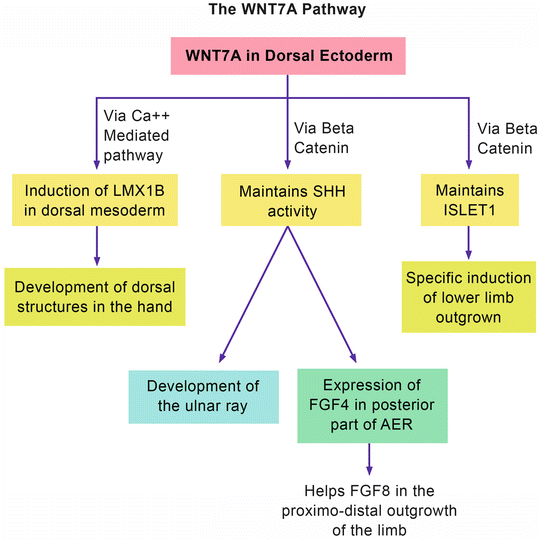
Fig. 12.4
The WNT7A pathway
Looking at the WNT7A pathway, one can speculate the phenotypes of syndromes associated with loss-of-function mutations of WNT7A: (1) the loss of LMX1B will result in ventralization of the dorsum of the hand; (2) the loss of SHH activity will result in a variable degree of ulnar ray deficiency as well as short upper limbs; and (3) the loss of ISLET1 maintenance will result in truncated lower limbs and pelvic dysplasia. The end result is a triad of ventral dimelia, ulnar ray deficiency, and truncated lower limbs.
Ventral Dimelia in Experimental Animals
Parr and McMahon [32] studied the effects of loss of function of Wnt7a in mice. The knockout mouse models showed ventral dimelia and ulnar ray deficiency but without truncation of the hind limbs. This may indicate that Wnt7A (beta-catenin)–ISLET 1 interactions are more functional in humans than mice.
Ventral Dimelia in Humans
Al-Qattan [10] classified ventral dimelia into three groups according to the severity of the phenotype. The classification was supported by the genetic basis of each group. The mildest phenotype is the nail-patella syndrome that is caused by LMX1B mutations. Features of nail-patella syndrome include hypoplastic/aplastic nails, absent patella, and renal defects. The second group has partial loss-of-function mutations of WNT7A leading the triad of ventral dimelia, mild ulnar ray deficiency, and truncated lower limbs. In the genetics literature, this is known as Fuhrmann syndrome. Two WNT7A mutations are known to be associated with the Fuhrmann phenotype: the R222W [33] and the A109T [34] mutations. The third group has complete loss of function of WNT7A; and as expected, this group has the most severe phenotype: severe ventral dimelia, severe ulnar ray deficiency, and frequently absent lower limbs. In the genetics literature, this severe phenotype is known as Al-Awadi syndrome. Three WNT7A mutations are known to be associated with Al-Awadi syndrome: the E72K [35], the R292C [34], and the G204S [36, 37] mutations (Fig. 12.5).