For diagnosis
Clinical evaluation
Biopsy rarely needed for confirmation
Genetic screen for mucopolysaccharidoses in extensive cases
For treatment
Tincture of time—most lesions resolve by age 6 years
The most common hyperpigmentation is the Mongolian spot (Fig. 11.1), which can be noted at birth or shortly thereafter [3]. The Mongolian spot is a bluish coloration noted with immature pigment cell placement in the skin, usually in infants of color who are full term and full-size. The bluish color comes from the Tyndall effect, an optical alteration noted when melanocytes are in the dermis. The leading location is the sacrum and gluteal region, but eccentric placement can be seen anywhere on the body. The Mongolian spot is seen in 9.5–18.9 % of Caucasian infants, 46 % of Hispanic/Latino infants, 62.2 % of Indian infants, 83.6 % of East Asians, and 96 % of Black newborns [4, 5]. Extensive lesions can be associated with mucopolysaccharidoses and may need genetic screen [6].
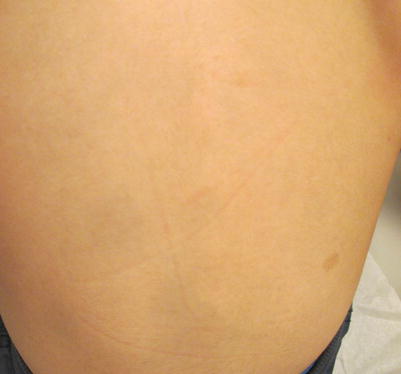
Fig. 11.1
Mongolian spot on the lower back of an 8-year-old Asian boy with a solitary café au lait macule off to the right side of the back
Most Mongolian spots will disappear with time, and no therapy is needed [7].
Café au Lait Macules
Café au lait macules (Fig. 11.1) are common localized areas of excess pigmentation that are generally round or oval, but may follow a segmental pattern. Although the classic lesion seen in light-skinned individuals or in association with neurofibromatosis is the color of light coffee, lesions can vary from nearly imperceptible in Caucasian infants to dark brown in Black children. It is estimated that 22 % of Black children and 11 % of Caucasian children have a solitary lesion. The lesions are most concerning when found in larger number [8].
The presence of six or more café au lait macules of 5 mm or greater in childhood or 1.5 cm in adulthood is a criterion for the diagnosis of neurofibromatosis type I, a multi-system autosomal dominant genetic disorder associated with tumor development [9]. Of 44 children in a neurofibromatosis type I clinic who had 6 or more café au lait at presentation, 34 went on to meet criteria for the disease—32 of whom met criteria by 72 months [10]. The vast majority of patients will be diagnosed by age 8 years, and 46 % of spontaneous cases by age 1 year [9].
Large segmental café au lait over the trunk may be associated with McCune Albright Disease seen with macrocephaly and precocious puberty. In the setting of multiple café au lait macules, clinical evaluation for the other criteria of neurofibromatosis includes an ophthalmology examination every 6 months, dermatologic evaluation every 6 months, and neurological, orthopedic and/or genetic screen where appropriate. Criteria for neurofibromatosis type 1 and the average timing of onset are listed in Table 11.1 [9].
Table 11.1
Diagnostic criteria for neurofibromatosis type I
Criterion | Clinical appearance | Timing of onset |
|---|---|---|
Café au lait | Six or more tan to brown ovoid macules and patches | Most children will meet this criteria by age 6 years |
>5 mm childhood | ||
>1.5 cm adulthood | ||
Optic nerve gliomas | Tumor of the optic nerve visualized on dilated examination | 10 % by age 3 years |
Lisch nodules (smooth muscle hamartomas of the iris) | Pigmented papules on the iris visualized with slit lamp examination | Age 6 or later; 68 % by age 10 years |
Osseous lesions | Pseudoarthrosis, sphenoid wing dysplasia, and dysplastic vertebrae seen on x-rays | 30 % at birth |
Relative | First degree relative with neurofibromatosis | About half are sporadic and half have a first-degree relative |
Neurofibromas | Two or more neurofibromas, which are soft tumors that indent with pressure, or plexiform neuroma, which is described as a bag of worms | Meet criteria: |
10 % at birth | ||
48 % by age 10 years | ||
84 % of 20 year olds | ||
Freckling | Pinpoint tan macules of the axillary and or inguinal folds | 90 % by age 6 years; rare after age 7 years |
For diagnosis |
Clinical confirmation/ examination (benign vs. in the setting of a systemic genetic syndrome) |
Biopsy in atypical lesions may help |
Full body skin examination every 6 months |
Ophthalmology examination every 6 months |
For treatment |
Laser may reduce appearance in some cases |
Excision of tumors that are causing functional impairment in the setting of neurofibromatosis type 1 |
Developmental and neurological evaluation in multiple lesions |
Nevus Depigmentosus (Fig. 11.2)
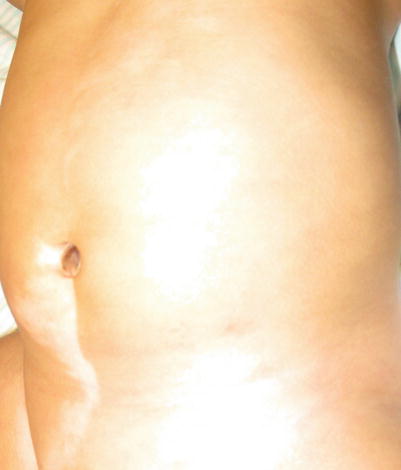
Fig. 11.2
Nevus depigmentosus on the lower abdomen in a Hispanic male infant
This misnomer represents a localized reduction in pigmentation seen at birth in children of Asian, Indian, and Hispanic/Latino descent [11]. The areas represent a localized increase in the ratio of pheomelanin to eumelanin and reduced melanosomes (melanin storage organelles). Fifty percent will be seen at birth and almost all lesions are noted by age 1 year.
Wood’s lamp examination will demonstrate partial highlighting and better define lesions. Nevus depigmentosus needs no therapy, but judicious sun protection to avoid enhancement of the color differential from normal skin is cosmetically beneficial [12].
For diagnosis |
Wood’s lamp equivocal |
Biopsy for hematoxylin and eosin stain or electron microscopy |
In cases of multiple lesions, full-body exam every 6 months with Wood’s lamp for highlighting |
Ophthalmic examination every 6 months |
For treatment |
No therapy needed. Most cases cause limited cosmetic disfigurement |
Cosmetic camouflage |
Tuberous Sclerosis
Multiple areas of hypopigmentation can be noted in systemic genetic disease such as tuberous sclerosis, presenting early on with confetti macules and thumbprint macules, and later with the botanically reminiscent ash leaf macules. The current criteria for tuberous sclerosis are listed in Table 11.2. Diagnosis requires ophthalmologic, neurologic, and genetic screens accompanied by frequent dermatologic follow-up (every 6 months) and imaging of the head, which may identify UBOs (unidentified bright objects) or intracranial tubers [13–16].
Table 11.2
Diagnostic criteria for tuberous sclerosis (two major or one major and two minor)
Criteria | Dermatologic clinical appearance | Frequency |
|---|---|---|
Major | ||
Angiofibromas or Fibrous forehead plaque | Flesh-colored papules perinasal | 32.1–100 % |
Similar to Shagreen but facial forehead localization (Both begin in toddler to early childhood years) | 2.5–55 % | |
Hypomelanotic macules (≥3, at least 5-mm diameter) | Ovoid hypopigmented lesions, usually truncal | 55.5–95 % |
Ungual fibromas (> or =2) | Flesh-colored to brown narrow spicules of skin originating in the cuticle and overhanging the nail | 20 % overall; 80 % of older patients |
Shagreen patch | Thick or indurated flesh-colored plaque, usually lower back; onset first decade | 12.3–83 % |
Multiple retinal hamartoma | Noted on ophthalmology exam | 30–50 % |
Cortical dysplasias | Tubers and cerebral white matter migration lines | |
Subependymal Nodules | Noted on brain scans; tubers and cerebral white matter migration lines | |
Subependymal giant cell astrocytoma | Noted on brain scans | |
Cardiac Rhabdomyoma | Noted in utero/at birth on ultrasound/echocardiography; self-resolving | 12 % have cardiac issues as a result |
Lymphangioleiomyomatosis (LAM) | Noted within the bones and lungs of the chest cavity with imaging | 38 % adults |
Angiomyolipomas | Noted mostly within the kidneys on imaging | |
Minor “Confetti” skin lesions | Scattered hypopigmented areas a few mm wide across the back | 3–58 % |
Dental enamel pits (>3) | Noted on dental evaluation | 100 % |
Intraoral fibromas (> or =2) | Noted on dental evaluation | 20–50 % |
Retinal achromic patch | 1.2–15 % | |
Multiple renal cysts | 6.2–28 % | |
Non renal hamartomas | ||
For diagnosis of tuberous sclerosis | |
Criteria evaluation, coordinated multidisciplinary evaluation—ophthalmology, dermatology, neurology, renal, pulmonary, genetics, dental, and cardiology | |
Skin and oral evaluation every 3–6 months | |
Biopsy for confirmation of growths which are not clinically typical, including fibrous forehead plaque | |
In cases of multiple lesions, full-body exam every 6 months with Wood’s lamp for highlighting | |
Chest X ray, EKG, echocardiography, abdominal ultrasound, CT scan head, MRI abdomen | |
Ophthalmic examination every 6 months | |
For treatment | |
Level of evidence | |
Coordinated multidisciplinary care—ophthalmology, dermatology, neurology, renal, pulmonary, genetics | |
Cosmetic camouflage | |
Cosmetic therapies are available to reduce facial lesions, including topical rapamycin | C |
Topical rapamycin for facial angiofibromas | C |
MTOR inhibitors (e.g. sirolimus) | A |
Nevoid Melanosis or Blaschkoid Pigmentation
Nevoid hypermelanosis or hypomelanosis (Fig. 11.3) (synonyms hypomelanosis of Ito, linear and whorled nevoid hypermelanosis, nevus depigmentosus, cutis tricolor or Blaschkoid dyspigmentation) is the appearance of tan to brown (slightly hypopigmented to hyperpigmented) lesions in a Blaschkoid pattern, usually on limbs with trunk and abdomen trailing as locations [17]. A Blaschkoid pattern means they follow the Lines of Blaschko, which are noted at birth or within the first 2 years of life, and represent localized cutaneous mosaicism in association with either a normal child (majority of cases) or neurologic (e.g. seizure and mental retardation), skeletal and multi-system abnormalities. When hypopigmented streaks are paired with neurologic abnormalities, the complex is often termed hypomelanosis of Ito. In two chart reviews, of 36 patients and 54 patients, only 13.9 % and 30 % of patients had extracutaneous manifestations, respectively [17, 18]. The majority of patients with Blaschkoid pigmentation were otherwise normal.
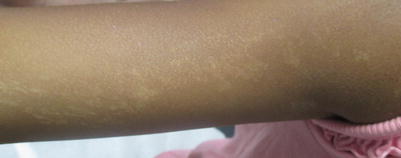
Fig. 11.3
Congenital hypopigmentation along the lines of Blaschko
Neurologic and ophthalmologic screen is indicated [19]. Gross skeletal evaluation of children with Blaschkoid dyspigmentation and limb measurement (assessment for symmetry or anomaly) and appropriate orthopedic evaluation where indicated. Genetic testing for chimerism is reasonable, with cutaneous genotyping of the two different tones of skin (from biopsy) being an optional screen for mosaicism, though unnecessary in the general setting. Where the underlying diagnosis is hard to establish, biopsy can be performed.
For diagnosis of syndromic cases |
Repeat cutaneous evaluation every 6–12 months in the first few years of life with Wood’s lamp for highlighting looking for anomalies, localized alterations in hair growth, overgrowth or atrophy of limb and associated cutaneous changes (e.g. palmoplantar keratoderma) |
Referrals to ophthalmology, neurology and genetics |
Biopsy for confirmation of diagnosis where uncertain |
Limb lengths and widths to identify underlying skeletal abnormalities |
For treatment |
Coordinated multidisciplinary care—ophthalmology, dermatology, neurology, genetics |
Cosmetic camouflage is available for visible lesions |
Laser therapies may be tried for hyperpigmented lesions similar to those used in the setting of café au lait |
Becker’s Nevus
The Becker’s Nevus (Fig. 11.4) is an organoid nevus that appears in a checkerboard mosaic pattern [20]. A smooth muscle hamartoma is usually accompanied by overlying hyperpigmentation, appearing like a café au lait and hypertrichosis. The constellation of findings can be seen in the infantile smooth muscle hamartoma, or appear around puberty on the trunk or proximal extremities, at which time it is usually termed a Becker’s Nevus. The younger the child, the less likely it is that hypertrichosis will be noted [21]. The Becker’s Nevus is felt to have estrogen receptors. It may be familial in nature [20]. When it is accompanied by hemimaxillary enlargement, asymmetry of the face, tooth abnormalities, and skin findings, it is termed the HATS association [22]. A rare association with neurofibromatosis type I has been reported [23]. Becker’s Nevus Syndrome is the presence of the Becker’s Nevus with unilateral breast hypoplasia and skin, muscle, or skeletal abnormalities. Mental retardation may be noted [24].
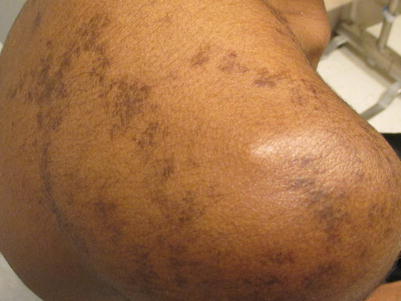
Fig. 11.4
Becker’s nevus on the shoulder in an African American male
The Becker’s Nevus can be diagnosed clinically in most cases. Confirmatory tests include Wood’s lamp to define the extent of pigmentation (especially needed for light-skinned patients or females with minimal, light body hair). Confirmatory biopsy can be performed demonstrating typical histology [21]. Imaging, dental, and orthopedic evaluations can aid in the defining of associated structural abnormalities, if suspected. A solitary report of a melanoma in a Becker’s Nevus has appeared in the literature, therefore biopsy of changing lesions or for the appearance of new lumps or bumps is needed [25]. Removal of hair or pigmentation overlying a Becker’s nevus has been described, with variable success, using variable lasers, as well as fractional resurfacing laser [26, 27].
For diagnosis |
Clinical evaluation |
Biopsy for confirmation |
Chest X-ray, Dental X-rays, and MRI can define underlying defects |
For treatment |
Biopsy of alterations in pigmentation, lumps, bumps, or nodules |
Cosmetic camouflage |
Laser therapies have been described including hair removal, pigment removal lasers, and fractional resurfacing |
Nevus of Ito and Nevus of Ota
These melanocytic nevi are named based upon their localization in the skin [28]. Presenting in grey-blue sheet of color in the first year of life, they may affect any place in the head and neck region. The Nevus of Ota (Fig. 11.5) usually appears in the first or second trigeminal distribution, with associated ocular component in some cases, while Nevus of Ito appears in the shoulder girdle. In these cases, ongoing ocular assessment is needed to identify associated glaucoma and/or melanoma of the eye. In the skin, palpation for lumps and bumps and ongoing cutaneous evaluation is recommended. Because of the rare association with meningeal melanocytomas which need to be excised, observation for neurologic changes (e.g. headaches) is needed [29, 30]. The lesions are more common in Asians and may cause stigma [28]. A variety of laser techniques for removal are available with variable success [31]. These lesions, in contrast to the Mongolian Spot, do not fade with time.
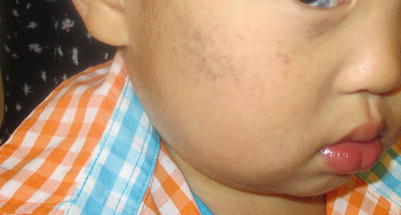
Fig. 11.5
Nevus of Ota in an Asian male infant
Congenital Widespread Alterations in Pigmentation
Oculocutaneous Albinism
Clinical Features
Oculocutaneous albinism (OCA) results from gene defects in the melanin synthesis pathway and affects 1:20,000 [32]. The specific defective gene determines if patients will have either decreased melanin synthesis or complete absence of melanin synthesis [32–34]. The adverse effect on melanin is manifested in hair follicles, ocular structures, and the skin. OCA types 1 through 4 have normally structured melanocytes, but the melanocytes are unable to make pigment. Patients with OCA Type I have pink nevi, severe nystagmus, and increased risk of squamous cell carcinomas. OCA Type 1A has completely absent tyrosinase, and thus begets the most severe clinical manifestations and highest skin cancer risk. In OCA Type 1B, the tyrosinase is present, but decreased, producing clinical variants such as yellow mutant. OCA Type 2 results from a defect in P protein, and results in the brown variant where those afflicted have nystagmus, light brown hair, and pigmented nevi, and may make more pigment as they get older. OCA Type 3 is due to a defect in tyrosine-related protein resulting in the Rufous subtype, where patients have nystagmus, blue or brown iris, light brown to red hair, and light brown-red-bronze skin. OCA Type 4 is due to mutation in membrane-associated transporter protein (MATP), a melanosome membrane transporter (Table 11.3). There are other hypomelanosis disorders that have oculocutaneous manifestations along with systemic manifestations, including Hermansky-Pudlak syndrome, Chediak-Higashi syndrome and Griscelli syndrome.
Table 11.3
Types of oculocutaneous albinism (OCA)
Type of OCA | Inheritance | Gene defect and pathogenesis | Distinguishing clinical features |
|---|---|---|---|
Type 1a (Tyrosine-negative) | AR | TYR, tyrosinase enzyme deficiency; absence of tyrosinase activity or inability tyrosinase to transport to melanosomes | Absence of melanin in skin, hair, and eyes |
White hair may become slightly yellow over time | |||
Severe nystagmus | |||
Amelanotic nevi (pink nevi) | |||
Blue-gray eyes, decreased visual acuity, prominent red reflex throughout life | |||
Increased risk of squamous cell carcinomas | |||
Type 1b (yellow mutant) | AR | TYR, tyrosinase is present but decreased | Develops some pigment over time |
Type 2 (Tyrosinase positive) (brown variant) | AR | P gene, defect in P protein, decreased eumelanin synthesis, normal number of melanocytes | Brown variant is most common form of albinism in those of African descent |
Light brown hair | |||
Pigmented nevi | |||
Eye symptoms may improve with age | |||
May get more pigment with increased age | |||
Type 3 (Rufous) | AR | TYRP-1, Tyrosinase-related protein 1 | Nystagmus |
Decreased visual acuity | |||
Blue-brown iris | |||
Light brown-red-bronze skin | |||
Type 4 | MATP, Mutation in membrane-associated transporter protein, a melanosomes membrane transporter | Similar to OCA type 2 |
Investigations Recommended
For diagnosis |
Biopsy to determine presence of melanocytes or not (may be required) |
Family history (required) |
Genetic testing |
Ophthalmologic examination for nystagmus, strabismus, photophobia, poor vision |
For on-going management |
Dermatology follow up to biopsy any suspicious lesions to rule out skin cancer (Basal cell carcinoma, Squamous cell carcinoma, melanoma) |
Ophthalmologic follow-up |
Sun avoidance, sunscreen, eye protection, protective clothing |
Phenylketonuria
Clinical Features
Phenylketonuria (PKU) results from a deficiency of phenylalanine hydroxylase (PAH) or tetrahydrobiopterin, the phenylalanine hydroxylase co-factor [35, 36]. PKU is inherited via autosomal recessive pattern and affects 1:10,000–15,000 Caucasian newborns. The PAH gene is found on chromosome 12q23.1, but there are over 600 reported mutations. Patients with similar PAH defects may have different phenotypes. Without functioning PAH, the pathway of phenylalanine-to-tyrosine-to-dopamine-to-melanin does not occur. Tyrosine is needed for production of melanin, which is necessary for hair and skin color.
In addition to skin hypopigmentation, mental retardation, dermatitis, photosensitivity, and seizures may occur. Most patients have fair skin, blonde hair, and blue eyes. A musty odor is often noted from the by-products (phenylacetic acid) of the failed metabolic pathway. Patients with high levels of phenylalanine may have atopic dermatitis-like dermatitis and sclerodermoid-like skin changes, which improve with lowered phenylalanine levels.
Screening for PKU is a component of many state-mandated newborn screening programs [35–38]. If recognized early, a phenylalanine-free formula can be instituted, and later a low-phenylalanine diet. The dietary restrictions are instituted to prevent the most severe adverse events and clinical features that result from high phenylalanine levels. Despite these strict diet-control efforts, neurodevelopmental and psychological problems may still emerge.
Investigations Recommended
For diagnosis
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|