Abstract
The term cutaneous T-cell lymphoma (CTCL) describes a heterogeneous group of neoplasms of skin-homing T cells that show considerable variation in clinical presentation, histologic appearance, immunophenotype and prognosis. CTCLs represent approximately 75–80% of all primary cutaneous lymphomas, whereas primary cutaneous B-cell lymphomas (CBCLs) account for approximately 20–25%. For many years, mycosis fungoides (MF) and Sézary syndrome (SS) were the only known types of CTCL. Over the past two to three decades, based on a combination of clinical, histologic, immunophenotypical and genetic criteria, new types of CTCL have been defined and included in new classifications for (primary cutaneous) malignant lymphomas, such as the WHO-EORTC classification for cutaneous lymphomas and the fourth edition of the WHO classification for malignant lymphomas (2008, updated in 2016). In these classifications, three main categories of CTCL can be distinguished: (1) the group of classic CTCL, which includes MF, variants or subtypes of MF, and SS; (2) the group of primary cutaneous CD30-positive lymphoproliferative disorders (CD30+ LPD), which includes lymphomatoid papulosis and cutaneous anaplastic large cell lymphoma; and (3) a group of rare, often aggressive cutaneous T/NK-cell lymphomas, including subcutaneous panniculitis-like T-cell lymphoma, extranodal NK/T-cell lymphoma, nasal type, and cutaneous peripheral T-cell lymphoma (PTCL), NOS (not otherwise specified). In this latter group, aggressive epidermotropic CD8-positive CTCL, cutaneous gamma/delta T-cell lymphoma, primary cutaneous CD4-positive small-medium pleomorphic T-cell lymphoproliferative disorder and primary cutaneous acral CD8-positive lymphoma (added in the 2016 update of the WHO classification) have been separated out as rare subtypes of PTCL, NOS.
Classification according to the WHO-EORTC classification or WHO 2008/2016 classification is the most important prognostic factor and a prerequisite for adequate management and treatment of these conditions. This chapter describes the characteristic features, differential diagnosis, management and treatment, and prognostic factors of these different types of CTCL.
Keywords
cutaneous T-cell lymphoma, mycosis fungoides, Sézary syndrome, primary cutaneous CD30-positive lymphoproliferative disorders, subcutaneous panniculitis-like T-cell lymphoma, adult T-cell leukemia/lymphoma, extranodal NK/T-cell lymphoma (nasal type), primary cutaneous CD8-positive aggressive epidermotropic cytotoxic T-cell lymphoma, primary cutaneous gamma/delta T-cell lymphoma, primary cutaneous CD4-positive small/medium T-cell lymphoproliferative disorder, primary cutaneous acral CD8-positive T-cell lymphoma, folliculotropic mycosis fungoides, pagetoid reticulosis, granulomatous slack skin
Introduction
The term cutaneous T-cell lymphoma (CTCL) describes a heterogeneous group of neoplasms of skin-homing T cells that show considerable variation in clinical presentation, histologic appearance, immunophenotype, and prognosis. CTCLs represent approximately 75–80% of all primary cutaneous lymphomas, whereas primary cutaneous B-cell lymphomas (CBCLs) account for approximately 20–25% . For many years, mycosis fungoides (MF) and Sézary syndrome (SS) were the only known types of CTCL. Over the past three decades, based on a combination of clinical, histologic and immunophenotypical criteria, new types of CTCL and CBCL have been defined and new classifications for the group of primary cutaneous lymphomas have been formulated . A major advantage in the management of primary cutaneous lymphomas compared with lymphomas arising at other sites is that the former can be seen and can be biopsied easily, giving the dermatologist the unique opportunity to correlate the clinical appearance and clinical behavior with histologic, immunophenotypical, and genetic aspects of these conditions. Hence, the dermatologist can play a key role in the diagnosis, classification, and treatment of these diseases.
History
In 1806, a French physician, Jean Louis Alibert, was the first to describe a patient with MF. This case was designated pian fungoïde in his atlas, but in 1835 it was renamed mycosis fungoïde because of the resemblance of some skin tumors to mushrooms. In 1870, Bazin described the natural progression from a nonspecific premycotic phase to plaque lesions and finally to tumors, which probably represents one of the first descriptions of the “multistep model” in the development of a malignancy. In 1885, Vidal and Brocq used the term “mycosis fungoides d’emblée” for patients presenting with skin tumors not preceded by patches or plaques. It has now become clear that such cases represent another type of CTCL or a CBCL. The erythrodermic form of MF was described in 1892 by Besnier and Hallopeau. These early descriptions of the major clinical forms of MF were followed by descriptions of Sézary syndrome by Sézary and Bouvrain in 1938, pagetoid reticulosis by Woringer and Kolopp in 1939, and lymphomatoid papulosis (LyP) by Macaulay in 1968.
Thus, in the early 1970s, MF and some related conditions were the only types of cutaneous lymphoma that had been rather well described. Reports on cutaneous lymphomas other than MF/SS, commonly designated in the past as malignant reticulosis or reticulum cell sarcoma, were few. Moreover, they were firmly believed to represent skin manifestations of a systemic lymphoma and treated as such.
The CTCL concept
In 1968, Lutzner and Jordan described the ultrastructural features of the circulating atypical cells in SS. Characteristically, the nuclei of these cells had deep and narrow indentations, giving them a cerebriform appearance. Three years later, similar cells were found in the skin and lymph nodes of patients with MF. In 1975, based on the observation that the neoplastic cells in MF, SS, and related conditions not only had the same morphology but also a common T-cell phenotype, the term “CTCL” was suggested for this group of diseases . Within a short time, this term gained wide acceptance, particularly in the US. The introduction of the CTCL concept can be considered as a landmark in the history of this group of diseases. However, a major disadvantage has been that in many subsequent studies, distinction was no longer made between MF, SS, and other T-cell neoplasms, entities which may vary considerably in clinical presentation and clinical behavior.
The concept of primary cutaneous lymphomas
At about the same time that the CTCL concept was introduced, several European groups started to classify cutaneous lymphomas according to the criteria of the Kiel classification, a system used by hematopathologists for the categorization of nodal lymphomas. It was then determined that many types of CBCL and CTCL (other than classic MF and SS) can present in the skin without any evidence of extracutaneous disease at the time of diagnosis. It appeared that these primary cutaneous lymphomas often have a completely different clinical behavior and prognosis when compared to morphologically similar lymphomas arising within lymph nodes, and therefore require different types of treatment . In addition, differences in the presence of specific chromosomal translocations and in the expression of oncogenes, viral sequences or antigens (e.g. EBV), and adhesion receptors involved in tissue-related lymphocyte homing were found.
Such differences underscored that primary cutaneous lymphomas represent a distinct group and may explain, at least in part, their different clinical behavior. For instance, the observation that the neoplastic T cells in most CTCLs express cutaneous lymphocyte antigen (CLA) and the CC-chemokine receptors 4 (CCR4) and 10 (CCR10) indicates that they are the neoplastic counterparts of normal skin-homing T cells, and this explains why these CTCLs present in the skin. Perhaps most importantly, it appeared that different types of CTCL and CBCL with different clinical behaviors and different therapeutic requirements may have an identical histologic appearance. This implies that histologic features should always be combined with clinical and immunophenotypical data, before a definite diagnosis (classification) is made.
Over the past two decades, such an approach resulted in the delineation of several new types of CTCL and CBCL, and it formed the basis of the European Organization for Research and Treatment of Cancer (EORTC) classification for primary cutaneous lymphomas .
EORTC, WHO and WHO-EORTC classification schemes
The EORTC classification scheme was the first one that was specifically designed for the group of primary cutaneous lymphomas. It contained a limited number of well-defined types of CTCL and CBCL, which together accounted for >95% of all primary cutaneous lymphomas, in addition to some provisional entities, which were not yet fully defined clinically. A distinction was made between cutaneous lymphomas with indolent, intermediate, or aggressive clinical behavior. By including well-defined and recognizable disease entities, this classification provided the clinician with detailed information on staging, preferred mode of treatment, clinical behavior and prognosis, serving as a useful guide to optimal management and treatment.
The third edition of the World Health Organization (WHO) classification for malignant lymphomas adopted the most common types of CTCL from the EORTC classification, but there was controversy with regard to the categorization of some rare types of CTCL . In addition, the WHO classification contained new entities not yet included in the EORTC scheme. In 2004, representatives from both organizations reached an agreement on a new classification scheme for the group of cutaneous lymphomas: the WHO-EORTC classification . Importantly, the fourth edition of the WHO classification for malignant lymphomas, published in 2008 (updated in 2016) and used by pathologists and hematologists worldwide, has adopted the terminology and definitions of the WHO-EORTC classification; therefore, it now includes all the types of CTCL and CBCL included in previous classifications of primary cutaneous lymphomas . The frequency and survival of patients with the different types of CTCL recognized in the WHO-EORTC classification are presented in Table 120.1 . Following a description of practical guidelines for diagnosis, classification and staging, relevant features of the different types of CTCL included in this classification are presented.
| WHO-EORTC CLASSIFICATION FOR CUTANEOUS T-CELL LYMPHOMAS | ||
|---|---|---|
| WHO-EORTC classification | Frequency (%) * | 5-year survival rate (%) * |
| Indolent clinical behavior | ||
| Mycosis fungoides (MF) | 54 | 88 |
| Mycosis fungoides variants | ||
| 6 | 80 |
| 1 | 100 |
| <1 | 100 |
| Primary cutaneous CD30-positive lymphoproliferative disorders | ||
| 10 | 95 |
| 16 | 100 |
| Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) | 1 | 82 |
| Primary cutaneous CD4-positive small/medium T-cell lymphoproliferative disorder ¶ | 3 | 75 # |
| Primary cutaneous acral CD8-positive T-cell lymphoma ¶ | <1 | 100 |
| Aggressive clinical behavior | ||
| Sézary syndrome (SS) | 4 | 24 |
| Adult T-cell leukemia/lymphoma (ATLL) | NDA | NDA |
| Extranodal NK/T-cell lymphoma, nasal type | 1 | <5 |
| Primary cutaneous CD8-positive aggressive epidermotropic cytotoxic T-cell lymphoma ¶ | <1 | 18 |
| Primary cutaneous gamma/delta T-cell lymphoma (PCGD-TCL) | 1 | <5 |
| Primary cutaneous peripheral T-cell lymphoma (PTCL), NOS | 3 | 16 |
* Data are based on 1476 CTCL patients registered by the Dutch and Austrian Cutaneous Lymphoma Groups 3 .
# Virtually 100% for patients presenting with a solitary lesion.
¶ Provisional entities in the 2016 update of the WHO classification.
Practical Guidelines for Diagnosis, Classification and Staging of CTCL
Diagnosis
The first step in the evaluation of a patient suspected of having CTCL is to decide if it represents a lymphoma or a benign condition. Skin biopsies – preferably deep punch biopsies 4–6 mm wide or an excisional or incisional biopsy from the most representative skin lesions – should be performed. In the US, broad saucerizations are also done for suspected patch/plaque stage MF.
Since prior treatment with topical corticosteroids or PUVA may change the original histology profoundly, biopsies from untreated skin lesions are preferred. Even if an adequate biopsy specimen is obtained, a definite diagnosis is not always possible. First, several types of CTCL, such as MF, are often preceded for years by skin lesions that are neither clinically nor histologically diagnostic. The gradual progression from this prediagnostic phase to overt lymphoma explains why it has always been so difficult to reach a consensus on the minimal histologic criteria needed for an unequivocal diagnosis of MF . Because of the indolent clinical behavior of the disease in such patients, a conservative approach is justified. In most cases, repeated biopsies, when appropriate, will ultimately result in the correct diagnosis.
Secondly, atypical T-cell infiltrates are found not only in CTCL but also in reactive conditions, e.g. lymphomatoid drug eruptions (pseudo-T-cell lymphomas) . Therefore, a definite diagnosis should always be based on a combination of clinical, histologic and, in most cases, immunophenotypical criteria, and it may be supplemented with the results of gene rearrangement analysis .
Immunophenotyping
Immunohistochemical studies on paraffin or frozen sections using antibodies reactive with cell-surface or cytoplasmic molecules are extremely important in the diagnosis and classification of cutaneous lymphomas. Nowadays, antigen retrieval techniques allow immunophenotyping on formalin-fixed, paraffin-embedded tissue sections, eliminating the need for frozen sections. By using a panel of such antibodies (see Ch. 0 ), distinction can be made between neoplasms of T-cell, B-cell, NK-cell, and myeloid or monocytic origin. Within the group of CTCLs, such studies have contributed to the delineation of new subtypes and provided important diagnostic and prognostic criteria.
With respect to the diagnosis of CTCL, demonstration of an aberrant phenotype, i.e. loss of one or more T-cell-associated antigens, such as CD2, CD3, CD4 or CD5, by the neoplastic T cells, can be considered as an important additional criterion in establishing a definite diagnosis of CTCL . However, loss of CD7 expression, which may be caused by chronic T-cell stimulation and is commonly observed in benign dermatoses, is not a reliable marker.
T-cell receptor gene rearrangement analysis
T-cell receptor (TCR) gene rearrangement analysis, utilizing a standardized assay (e.g. BIOMED-2), may provide useful information for the diagnosis and staging of malignant lymphomas . However, caution is warranted in interpreting the results of such analyses. Clonal T-cell populations have been detected not only in skin lesions, lymph nodes, and peripheral blood of patients with CTCL and in cases of chronic dermatitis preceding MF (“clonal dermatitis”; see Ch. 9 ), but also in skin lesions of patients with apparently benign conditions, such as pityriasis lichenoides et varioliformis acuta, lichen planus, lichen sclerosus and some pseudolymphomas .
Demonstration of clonal T-cell populations can therefore not be used as an absolute criterion of malignancy, but should always be considered in conjunction with clinical and histologic features, which together remain the “gold standard”. If the clinical and histologic findings are not consistent, and an aberrant phenotype is not detected, a definite diagnosis of CTCL should not be made. In the future, high-throughput TCR sequencing may be used to enhance detection of T-cell clones in patients with CTCL and for distinguishing CTCL from benign inflammatory diseases .
Classification
Once a diagnosis of CTCL has been made, the type of CTCL should be determined. As noted previously, the histologic diagnosis is not the final diagnosis, which means that CTCL cannot be classified on the basis of histologic criteria alone. For instance, in the case of a skin biopsy showing a dermal infiltrate which is composed predominantly of CD30-positive large anaplastic or pleomorphic T cells, the histologic diagnosis will be (CD30-positive) anaplastic large cell lymphoma (ALCL). However, depending upon the clinical presentation, the definite diagnosis may be: a primary cutaneous ALCL; cutaneous involvement of a systemic ALCL; large cell transformation of MF with expression of CD30; or LyP (in cases of recurrent self-healing papules). Obviously, each of these conditions requires a different approach in terms of staging and treatment.
In Table 120.2 , the differential diagnosis of four histologic categories has been elaborated. This histologic subdivision into epidermotropic CTCL, CTCL with diffuse pleomorphic infiltrates, CD30-positive CTCL, and subcutaneous CTCL is neither complete nor meant as an alternative classification. It is only presented as an illustration of how histology should be combined with clinical and immunophenotypical data to arrive at a definite diagnosis.
| DIFFERENTIAL DIAGNOSIS OF COMMON HISTOLOGIC PATTERNS IN CUTANEOUS T-CELL LYMPHOMA | ||
|---|---|---|
| Histologic category | Differential diagnosis | Diagnostic criteria/clues |
| Epidermotropic CTCL (simulating or consistent with plaque stage MF) | Mycosis fungoides (MF) |
|
| Pagetoid reticulosis |
| |
| Lymphomatoid papulosis (LyP), type B or type D |
| |
| Primary cutaneous CD8-positive aggressive epidermotropic cytotoxic T-cell lymphoma |
| |
| Sézary syndrome |
| |
| Adult T-cell leukemia/lymphoma |
| |
| CTCL with diffuse pleomorphic infiltrates (CD30 − ) (simulation or consistent with tumor stage MF; few CD30 + tumor cells may be present) | Tumor stage MF |
|
| Primary cutaneous peripheral T-cell lymphoma, NOS |
| |
| Primary cutaneous CD4-positive small/medium T-cell lymphoproliferative disorder (and/or nodular pseudo-T-cell lymphoma [see text]) |
| |
| CD30-positive CTCL (see Fig. 120.10 ) | Lymphomatoid papulosis, type A/C |
|
| Primary cutaneous anaplastic large cell lymphoma (C-ALCL) |
| |
| Systemic ALCL with secondary skin involvement |
| |
| MF with blastic transformation |
| |
| CTCL with subcutaneous involvement | Subcutaneous panniculitis-like T-cell lymphoma |
|
| Other types of CTCL |
| |
Practical guidelines
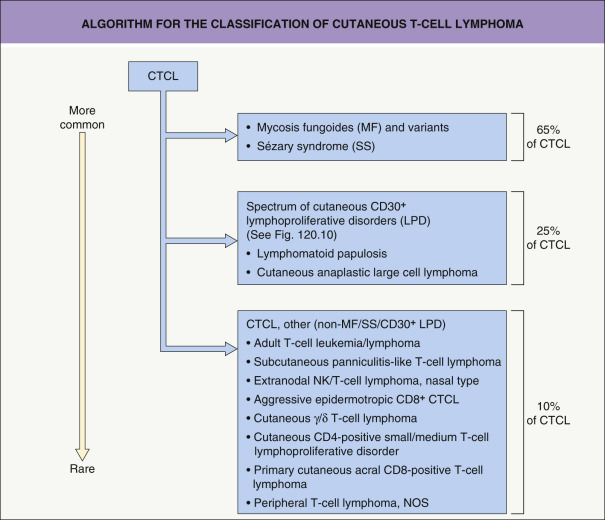
For those not regularly involved in the diagnosis and classification of CTCL and perhaps overwhelmed by the many new types of CTCL introduced in recent years, a stepwise approach, as presented in Fig. 120.1 , may serve as a practical guide .
Step one : Based on a combination of clinical, histologic and immunophenotypical criteria, distinction is first made between classic MF, MF variants, and SS on the one hand and CTCL other than these conditions on the other. The rationale for this first step is that dermatologists are familiar with the former set of conditions, which comprise approximately 65% of CTCLs, and these lymphomas require a different clinical approach in terms of staging and therapy compared to the other forms of CTCL.
Step two : The second category to be considered is the group of primary cutaneous CD30-positive lymphoproliferative disorders. It implies that evaluation of skin biopsies from patients with (suspected) CTCL should always include CD30 immunostaining. This group includes cases of cutaneous ALCL (C-ALCL) and LyP, which together represent the second most common group of CTCL (see Table 120.1 ). Patients within this spectrum of disease generally have an excellent prognosis, and most individuals can be managed easily by dermatologists.
Step three : With the first two steps, approximately 90% of CTCL will be classified correctly. The remaining group (10% of patients) contains rare types of CTCL, including subcutaneous panniculitis-like T-cell lymphoma (SPTCL), extranodal NK/T-cell lymphoma, nasal type, and the broad group of primary cutaneous peripheral T-cell lymphoma (PTCL), not otherwise specified (NOS). From the latter group, primary cutaneous CD8-positive aggressive epidermotropic cytotoxic T-cell lymphoma, primary cutaneous CD4-positive small/medium T-cell lymphoproliferative disorder, and primary cutaneous acral CD8-positive T-cell lymphoma have been separated out as provisional entities (see Table 120.1 ). In the WHO-EORTC classification, the term PTCL, NOS is maintained for remaining cases that do not fit into any of the provisional entities.
Apart from SPTCL, primary cutaneous CD4-positive small/medium T-cell lymphoproliferative disorder, and primary cutaneous acral CD8-positive T-cell lymphoma, lymphomas included in this third category have an aggressive clinical course, and they should be treated by, or in close collaboration with, a hemato-oncologist. It should be emphasized that because of overlapping clinicopathologic features and highly aberrant phenotypes, distinction between these different types of aggressive CTCL is sometimes very difficult. Distinction between primary and secondary cutaneous involvement is less important than in other types of cutaneous lymphoma in that patients presenting with only skin lesions generally develop extracutaneous disease within a short period of time and have a poor prognosis as well.
Staging
Apart from CTCL, a term preferably used only for primary CTCL, systemic T-cell lymphomas frequently present or relapse in the skin. Adequate staging procedures are required to differentiate between CTCL and these systemic lymphomas secondarily involving the skin. The extent of staging procedures is dependent on the type of (suspected) CTCL, and, in the case of classic MF, on the clinical stage of the disease . In cases of early patch/plaque stage MF, unequivocal cases of LyP, and probably pagetoid reticulosis as well, staging procedures are generally not worthwhile. In cases of suspected SS, adequate staging with special emphasis on assessment of peripheral blood involvement by cytology, immunophenotyping (flow cytometry), and TCR gene rearrangement analysis is essential. In all other types of CTCL, routine hematologic staging, including complete and differential blood cell counts, a serum chemistry panel, computed tomographic (CT) scans and/or positron emission tomography (PET) of the chest and abdomen, and bone marrow sampling, are required .
Mycosis Fungoides
Definition
Mycosis fungoides (MF) represents the most common type of CTCL and accounts for ~50% of all primary cutaneous lymphomas (see Table 120.1 ). The term MF should be restricted to the classic “Alibert–Bazin” type characterized by the typical evolution of patches, plaques and tumors, or for clinicopathologic variants showing a similar clinical course.
Epidemiology
MF has an incidence of about 0.4 per 100 000 inhabitants per year in the US . MF typically affects older adults (median age at diagnosis: 55–60 years), but it may occur in children and adolescents as well. Men are affected more often than women, with a male-to-female ratio of 1.6–2.0 : 1.
Pathogenesis
The etiology and the pathogenetic mechanisms involved in the development and stepwise progression of MF are largely unknown. Genetic, environmental, and immunologic factors have all been considered.
Genetic factors
Lymphomagenesis is considered to be a multifactorial process, in which a stepwise accumulation of genetic abnormalities may result in clonal proliferation, malignant transformation and, ultimately, progressive and widely disseminated disease. Although the successive clinical steps of tumor progression were described more than a century ago, the molecular events underlying the different steps of tumor progression have not been identified. Gene expression studies of early stage MF revealed overexpression of TOX, which may turn out to be a useful diagnostic marker . Several studies on tumor stage MF, using array-based comparative genomic hybridization, reported the same recurrent genetic aberrations including gains of chromosome 7p22–21 (45%), 7q21–22 (55%), 8q24 (35%) and 17q21 (40%) and losses of 9p21.3 (40%) and 13q14 (30%) . Loss of 9p21.3, which harbors the CDKN2A , CDKN2B and MTAP tumor suppressor genes, has been associated with a shorter survival in patients with tumor stage MF . In addition, constitutive activation of the NF-κB pathway in MF has been observed and may be explained in part by downregulation of NFKBIZ, an inhibitor of this pathway . Whole-genome DNA sequencing has also implicated NF-κB signaling in CTCL pathogenesis . Of note, somatic copy variants comprised 92% of driver mutations.
Environmental factors
Persistent antigenic stimulation has been demonstrated to play a crucial role in the development of various malignant lymphomas, including m ucosa- a ssociated l ymphoid t issue (MALT) lymphomas ( Helicobacter pylori infection), CBCL ( Borrelia burgdorferi infection), and enteropathy-type T-cell lymphoma (celiac disease). In MF, persistent antigenic stimulation has also been proposed as an initial event, but the nature of the antigen(s) involved is unknown. Large case–control studies have suggested a relationship with industrial or environmental exposures, but their role in the development of MF remains controversial . Whereas the etiologic roles of human T-cell leukemia virus 1 (human T-cell lymphotropic virus 1; HTLV-1) in adult T-cell leukemia/lymphoma and EBV in nasal NK/T-cell lymphoma have been firmly established, conclusive evidence for a primary etiologic role of these viruses in MF is lacking .
Immunologic factors
CD8 + cytotoxic T cells (CTL) are thought to play a crucial role in the antitumor response in MF. A relationship between high percentages of CD8 + CTL in the dermal infiltrates and improved survival has been described . These CD8 + T cells exert their antitumor effect both by a direct cytotoxic effect and by the production of cytokines, particularly interferon (IFN)-γ. They can mediate tumor cell lysis via exocytosis of cytotoxic granules containing perforin, granzymes and T-cell-restricted intracellular antigen (TIA-1), and by expression of Fas ligand (FasL), which interacts with Fas (CD95; APO-1) on the neoplastic T cells . Both pathways ultimately lead to activation of caspase 3 and tumor cell death. Loss of Fas expression or function by the neoplastic T cells is one of the many mechanisms by which tumor cells can escape from an effective antitumor response .
The neoplastic T cells in SS and tumor stage MF are derived from CD4 + T cells with a Th2 cytokine profile (production of IL-4, IL-5 and IL-10), whereas the cytotoxic T cells are the main producers of IFN-γ, which plays an important role in augmenting T-cell- and NK-cell-mediated killing. In accordance with this concept, a gradual shift from a predominantly type 1 cytokine profile in MF plaques to a predominantly type 2 cytokine profile in MF tumors has been suggested. Increased levels of Th2 cytokines may impair the Th1 cell-mediated antitumor response and contribute to the immunosuppression seen in patients with advanced MF .
Clinical features
Characteristically, patients with classic MF progress from patch stage to plaque stage and finally to tumor stage disease, and they have a protracted clinical course over years or even decades. Before a definite diagnosis is made, patients generally have many years of nonspecific eczematous or psoriasiform skin lesions and non-diagnostic biopsies. The median duration from onset of skin lesions to the diagnosis of MF is 4–6 years, but it may vary from several months to more than five decades .
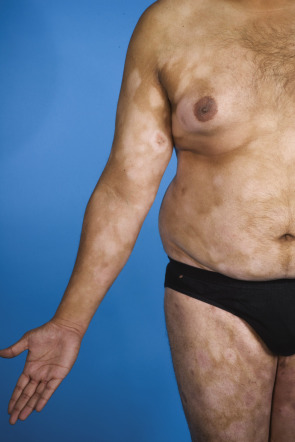
Early patch stage MF is characterized by the presence of variably sized erythematous, finely scaling lesions, which may be mildly pruritic ( Fig. 120.2A ). These early lesions may show variable degrees of atrophy, and a poikilodermatous variant consisting of patches with mottled hyper- and hypopigmentation, atrophy, and telangiectasia has been described (formerly called poikiloderma vasculare atrophicans). Generalized hypopigmented lesions may be observed in patients with darkly pigmented skin ( Fig. 120.3 ), and this is also a common presentation of juvenile-onset MF.
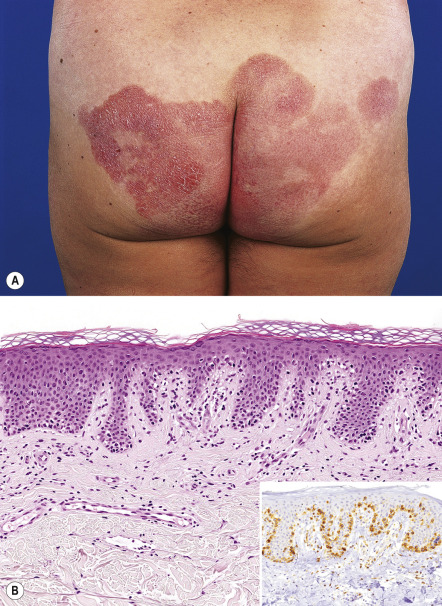
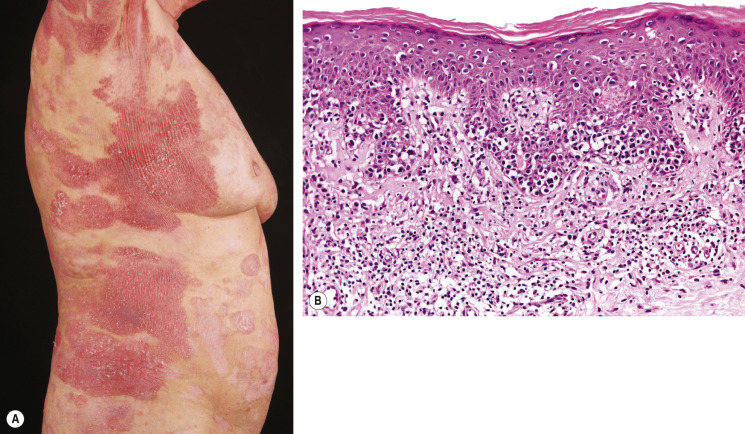
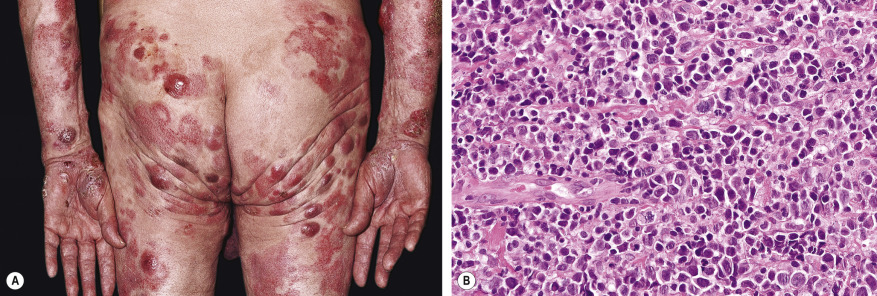
The initial skin lesions have a predilection for the buttocks and other covered sites of the trunk and limbs. With progression, more infiltrated reddish-brown, scaling plaques develop, which gradually enlarge and may have an annular, polycyclic or typical horseshoe-shaped configuration ( Fig. 120.4A ). It should be stressed that many patients never progress beyond the plaque stage of the disease. However, a minority (~10%) of patients may develop nodules or tumors. These patients with tumor stage MF characteristically show a combination of patches, plaques, and tumors ( Fig. 120.5A ); the latter often show ulceration.
If only skin tumors are present without preceding or concurrent patches or plaques, a diagnosis of MF is highly unlikely and another type of CTCL should be considered. The risk of developing extracutaneous disease correlates with the extent and type of skin lesions. It is exceedingly rare in patients with limited patch/plaque stage disease, relatively uncommon in patients with generalized plaques, and most likely in patients with skin tumors or erythroderma . Extracutaneous dissemination, almost without exception, first involves the regional lymph nodes draining areas of extensive skin involvement. Visceral involvement may develop subsequently and can involve any organ. The bone marrow is rarely involved.
Pathology
Early patch lesions in MF show superficial band-like or lichenoid infiltrates, consisting primarily of lymphocytes. Atypical cells, with small to medium-sized, highly convoluted (cerebriform) and sometimes hyperchromatic nuclei, are few in number and are mostly confined to the epidermis (epidermotropism). These lymphocytes characteristically colonize the basal layer of the epidermis as single cells surrounded by vacuolated halos, often in a linear configuration ( Fig. 120.2B ) .
In plaques , epidermotropism is generally more pronounced ( Fig. 120.4B ). The presence of intraepidermal nests of atypical cells (referred to as Pautrier microabscesses, although the initial description was by Darier) is a highly characteristic feature, but is observed in only a minority of cases. The epidermis may show acanthosis and elongated rete ridges, but spongiosis is generally mild or absent. The dermal infiltrates are also more pronounced, and they may contain a higher number of atypical cells with cerebriform nuclei and occasional blast cells, as well as admixed eosinophils and plasma cells. Rarely, a predominantly interstitial infiltrate that resembles granuloma annulare or morphea is observed (interstitial MF) .
With progression to tumor stage MF, the dermal infiltrates can involve the entire dermis and extend into the subcutaneous tissue. Epidermotropism may no longer be present. The tumor cells increase in number and size, showing variable proportions of small, medium-sized or large cells with cerebriform nuclei, blast cells with prominent nuclei, and intermediate forms ( Fig. 120.5B ). Large cell transformation , defined by the presence of CD30-negative or CD30-positive large cells exceeding 25% of the infiltrate or forming microscopic nodules, may occur and is generally associated with a poor prognosis. However, within this group, CD30-positive patients have a much better prognosis than CD30-negative patients .
Immunophenotype
The neoplastic cells in MF have a mature CD3 +, CD4 + , CD45RO + , CD8 − memory T-cell phenotype. In a minority of patients with otherwise classic MF, a CD3 + , CD4 − , CD8 + mature T-cell phenotype or more rarely a γ/δ T-cell phenotype (βF1 − , TCRγ + , CD3 + , CD4 − , CD8 + ) may be seen . Immunophenotyping may demonstrate (partial) loss of T-cell antigens in plaque or tumor stage MF, but rarely in early patch lesions, and is therefore of little help in early diagnosis.
Differential diagnosis
Regarding the differential diagnosis of MF, three categories should be considered. The first category contains a diverse group of benign dermatoses, which early MF may resemble clinically, and it includes several types of eczema, psoriasis, superficial fungal infections, and drug reactions. These specific diagnoses can generally be excluded by histologic and other standard dermatologic examinations. This category may also include patients with large plaque parapsoriasis (parapsoriasis en plaque) who show slightly scaly, sometimes atrophic, erythematous patches or plaques, which are commonly located on the trunk and buttocks (see Ch. 9 ). Whereas large plaque parapsoriasis cannot be distinguished clinically from early patch or plaque stage MF, the histologic features are often not consistent with MF. Long-term follow-up studies have documented progression of large plaque parapsoriasis to overt MF in ~10% of cases. However, one prevailing opinion is that large plaque parapsoriasis should be considered a form of MF, rather than a potential precursor of MF, but the author feels there is no consensus. There is even less consensus about whether small plaque parapsoriasis represents MF.
A second category includes several benign conditions with histologic features highly suggestive of MF. Examples of these are lymphomatoid contact dermatitis, lymphomatoid drug reactions, and actinic reticuloid. Apart from subtle histologic differences (e.g. the predominance of atypical T cells in the dermal infiltrates rather than in the epidermis), careful evaluation of the clinical features, which are generally not consistent with MF, often results in a correct diagnosis .
The third category includes other types of (epidermotropic) CTCL, which may resemble MF histologically. Diagnostic features of these entities are presented in Table 120.2 .
Staging systems and staging procedures
Staging patients with MF and SS is important, since it determines management and treatment and has prognostic significance. In 2007, a revised clinical staging system for MF and SS was proposed, which is based on the TNM (tumor–node–metastasis) classification system and takes into account the type and extent of skin lesions (T1–4) as well as the presence or absence of nodal (N0–3), visceral (M0–1), and peripheral blood involvement (B0–2) ( Tables 120.3 & 120.4 ) .
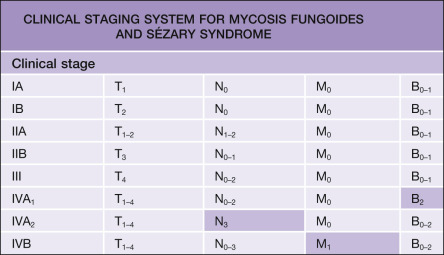
Evaluation of patients suspected of having MF should include a thorough physical examination with special attention to the type and extent of skin lesions and the presence of palpable lymph nodes, as well as skin biopsies, complete blood counts, and serum chemistries. Enlarged lymph nodes should be biopsied. Histologically, distinction can be made between lymph nodes showing dermatopathic lymphadenopathy without involvement by MF (N1), dermatopathic lymphadenopathy with early MF involvement (N2), and lymph nodes showing effacement of the normal lymph node architecture by neoplastic T cells (N3). The prognostic significance of such a subdivision has been well established.
No further examinations are recommended for patients with stage IA–B disease. CT scans of the chest and abdomen are recommended in patients in whom extracutaneous disease is suspected, but they are less useful in patients with limited patches and/or plaques without lymphadenopathy. Examination of other organs, including the bone marrow, should only be performed if clinically indicated.
Treatment
The choice of an initial treatment in MF depends on the stage of the disease and the general condition and age of the patient . Given the chronic and recurrent nature of MF, treatment should be aimed at improving symptoms while limiting toxicity. Following the traditional “stage-based” approach, skin-directed therapies are preferred in the early stages of MF (stages IA–IIA) and even in patients with limited tumor stage MF (IIB) ( Table 120.5 ). These skin-directed therapies include topical or intralesional corticosteroids, topical cytotoxic agents (e.g. mechlorethamine [nitrogen mustard]), phototherapy, and radiotherapy. The efficacy of skin-directed therapies in MF is explained by the preferential localization of the neoplastic skin-homing T cells to the epidermis and superficial dermis. Systemic multi-agent chemotherapy is not useful in these early stages, since it does not improve survival and is associated with considerable morbidity. In patients with refractory or progressive skin disease, skin-directed therapies can be combined with IFN-α or systemic retinoids. Alternatively, novel agents such as denileukin diftitox or h istone d e ac etylase i nhibitors (HDACi) such as vorinostat and romidepsin can be used, before systemic chemotherapy is considered. Both HDACi and denileukin diftitox have been approved in the US for patients with relapsed and refractory CTCL, but have not been registered for CTCL in Europe. In general, systemic chemotherapy is only indicated in advanced stages when there is nodal or visceral involvement or in patients with rapidly progressive tumors unresponsive to less aggressive therapies.
| TREATMENT OF MYCOSIS FUNGOIDES |
|---|
| Premycotic phase |
| Stage IA–IIA (patches/plaques) |
| Stage IIB (skin tumors) |
|
| Stage III (erythroderma) |
| Stage IV (nodal, visceral involvement) |
|
§ Address UV-shielded areas (e.g. upper inner thighs) via positioning.
^ Bexarotene 1% gel, tazarotene 0.1% gel, alitretinoin 0.1% gel.
* Efficacy compared to traditional treatments yet to be determined.
Skin-directed therapies
Topical corticosteroids
In many MF patients with only patches and thin plaques, application of topical corticosteroids is effective in controlling disease activity. In patients with limited patch/plaque stage disease (mainly patches), complete remissions in up to 60% of patients have been reported . In more advanced stages, they continue to be an important adjuvant therapy.
Topical chemotherapy
Topical application of mechlorethamine has proven to be an effective treatment for early stage MF. Mechlorethamine, either dissolved in water or compounded in an ointment- or gel-based preparation (see Ch. 129 ), results in complete remissions in approximately 60–80% of patients with stage IA–B disease . Most patients with early MF remain clear on maintenance therapy. Side effects include skin irritation, allergic contact dermatitis, and an increased risk for the development of skin cancer related to long-term use.
Radiotherapy
Total skin electron beam irradiation (TSEB) with an energy of 4–6 MeV is a highly effective treatment in patients with skin-limited MF (see Ch. 139 ) . The total dose is generally 36 Gy administered in fractions of 1.5–2 Gy over an 8–10-week period. Recently, lower total doses (10–12 Gy) have been employed, with the advantages of a shorter duration of treatment, fewer side effects, and opportunity for re-treatment . TSEB is most effective in patients with stage IA–B disease, with complete response rates of >80%. However, in most centers, such patients are treated with phototherapy or topical chemotherapy. TSEB is particularly useful in patients with tumor stage MF, where complete response rates of ~40% have been reported. Side effects are generally mild and include erythema, scaling, and temporary loss of hair, nails and sweat gland function.
Local radiotherapy with X-ray or preferably electron beam may be considered for single tumors in patients with plaque stage disease, either in combination with other modalities (e.g. PUVA) as an alternative for TSEB, or for new tumors following TSEB. A dose of ≥8 Gy suffices . In patients with unilesional MF, local radiotherapy may be curative.
Phototherapy
Several types of phototherapy can be used in the treatment of MF ( Ch. 134 ). These include PUVA therapy, broadband and narrowband UVB therapy, and, more recently, UVA1 therapy. Extracorporeal photopheresis (ECP) may be effective in patients with erythrodermic MF (see the section on Sézary syndrome ).
PUVA treatment has become a standard therapy for the early stages of MF . In patients with stages IA–IIA, complete response rates of 80–90% have been reported. In many centers, maintenance PUVA therapy (every 2 to 4 weeks) is given to prolong remission. Although sustained complete remissions have been reported, most patients will relapse after cessation of PUVA therapy or during maintenance treatment. Recurrent or persistent lesions particularly favor UV-shielded areas, such as the inner thighs and the gluteal cleft. In tumor stage MF, PUVA therapy alone is unlikely to result in complete responses, but favorable results may be achieved when combined with IFN-α, systemic retinoids or radiotherapy.
In patients with only patches, narrowband UVB therapy (311 nm) is increasingly being used as a safe and effective alternative to PUVA therapy with similar efficacy .
Systemic therapies (other than chemotherapy)
Interferons
The most commonly prescribed biological response modifier is interferon-alpha (IFN-α). In most centers, IFN-α is administered subcutaneously in doses of 3 to 9 million units three times a week. Side effects are generally mild and reversible, and they include flu-like symptoms, hair loss, nausea, depression, and bone marrow suppression. The overall response rate of IFN-α, when used as a single agent, is ~50%, with 17% representing complete remissions . The combination of PUVA and IFN-α appears to produce higher response rates than PUVA alone, and this combination may also be considered in patients with early tumor stage disease, when PUVA therapy alone is insufficient.
Retinoids
When used as a single agent, the overall and complete response rates of several first- and second-generation oral retinoids (isotretinoin, acitretin) and a novel RXR-selective retinoid (bexarotene) are roughly similar to those of IFN-α. A combination of retinoids (including bexarotene) plus PUVA (RePUVA) produces response rates similar to those of PUVA alone, although patients treated with RePUVA require fewer treatments and a lower cumulative UVA dose . In many centers, bexarotene has replaced the earlier-generation retinoids, but comparative studies have never been performed. In patients with patches or thin plaques, topical retinoids (bexarotene, tazarotene, alitretinoin) may be considered, but skin irritation is a limiting factor .
Denileukin diftitox
Denileukin diftitox is a fusion protein, in which diphtheria toxin is linked to IL-2. It binds to the high-affinity IL-2 receptor expressed by the neoplastic T cells in MF, and internalization of the toxin results in inhibition of protein synthesis and cell death. Overall and complete clinical response rates are ~30% and 10%, respectively . Denileukin diftitox can have substantial side effects, including capillary leak syndrome, fever, and fluid retention. However, at the time of writing, it is not commercially available.
Histone deacetylase inhibitors
HDACi, such as vorinostat and romidepsin, represent a novel class of drugs used in cancer therapy. Inhibition of the enzyme HDAC affects the expression of many genes (and their protein products) that are involved in cellular proliferation, differentiation, migration and apoptosis. Recent studies of both vorinostat and romidepsin report overall response rates of ~35% in patients with MF and SS, but complete responses are rare . The most common side effects are fatigue, gastrointestinal symptoms, and reversible thrombocytopenia. It remains to be determined which patients are most likely to benefit from this therapy.
Systemic chemotherapy
Systemic multi-agent chemotherapy should only be used in patients with unequivocal lymph node or visceral involvement, or in patients with progressive skin tumors that have failed to respond to other therapies. In many centers, the standard treatment in such cases is the administration of six cycles of CHOP ( c yclophosphamide, h ydroxydaunomycin [doxorubicin], O ncovin ® [vincristine] and p rednisone). With this and other combination regimens, high response rates can be achieved for extracutaneous involvement, but the responses are generally short-lived. Moreover, concurrent patches and plaques are often less responsive, and may require additional treatment with PUVA or mechlorethamine. Recent studies utilizing pentostatin, gemcitabine, and liposomal doxorubicin have demonstrated high overall response rates in patients with advanced MF, but controlled studies are still lacking .
In young patients with refractory and/or progressive MF and SS, an allogeneic hematopoietic stem cell transplantation may be considered. Using non-myeloablative reduced-intensity conditioning regimens, durable responses have been reported, but the optimal conditioning regimen and optimal timing for the transplant are still a matter of debate . Results with autologous hematopoietic stem cell transplantation in MF and SS have been disappointing , suggesting the need for a graft-versus-tumor response.
Prognosis
The prognosis of patients with MF is dependent on the stage, and in particular the type and extent of skin lesions and the presence of extracutaneous disease . Patients with limited patch/plaque stage MF have a similar long-term life expectancy as an age-, sex-, and race-matched control population. The disease-related 10-year survival is 96% for stage IA, 77–83% for stage IB, 42% for stage IIB, but only 20% for stage IV . Patients usually die of systemic involvement or infections.
Variants of Mycosis Fungoides
Apart from the classic Alibert–Bazin type of MF, many clinical and/or histologic variants have been reported. Clinical variants, such as bullous and hyper- or hypopigmented MF, have clinical behavior similar to that of classic MF, and they are therefore not considered separately. In contrast, folliculotropic MF, pagetoid reticulosis, and granulomatous slack skin have distinctive clinicopathologic features, and therefore they have been included as distinct variants or subtypes of MF in the WHO-EORTC classification scheme.
Folliculotropic MF
▪ Folliculocentric MF ▪ Pilotropic MF ▪ MF-associated follicular mucinosis ▪ Adnexotropic (folliculotropic and syringotropic)
Definition
Folliculotropic MF is a distinct variant of MF characterized by the presence of folliculotropic infiltrates, often with sparing of the epidermis and preferential involvement of the head and neck region. Most cases show mucinous degeneration of the hair follicles (follicular mucinosis; Ch. 46 ) and are traditionally designated as MF-associated follicular mucinosis. Similar cases, but without follicular mucinosis, have been reported as folliculocentric or pilotropic MF. From a biologic point of view, the most relevant feature (irrespective of the presence or absence of follicular mucinosis) is the deep, follicular and perifollicular localization of the neoplastic infiltrates, which makes them less accessible to skin-directed therapies. For both groups, the term folliculotropic MF is therefore preferred .
Epidemiology
This variant is found in approximately 10% of MF patients . It occurs mostly in adults, but may occasionally affect children and adolescents. Men are affected more often than women.
Clinical features
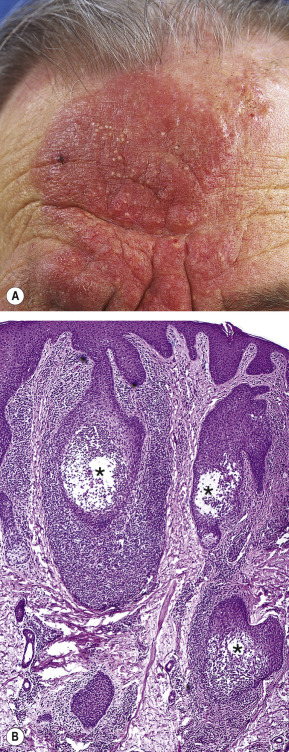
Patients may present with (grouped) follicular papules, acneiform lesions, indurated plaques and sometimes tumors, which preferentially involve and are most pronounced in the head and neck area (see Fig. 36.16 ). The skin lesions are often associated with alopecia, and sometimes with mucinorrhea. Infiltrated plaques in the eyebrow region with concurrent alopecia are a common and highly characteristic finding ( Fig. 120.6A ). Pruritus is often more severe than in classic MF and may represent a reliable parameter of disease activity. Secondary bacterial infections are frequently observed.
It should be stressed that the clinical staging systems for MF are not very helpful in patients with folliculotropic MF. Because of the perifollicular localization of the neoplastic infiltrates, many patients presenting with one or a few plaques on the face do not have stage IA disease, but rather should be considered as having tumor stage disease with a corresponding prognosis . However, more recent studies have reported that patients presenting with only follicular papules or very thin plaques may have an excellent prognosis similar to that of early-stage classic MF .
Pathology
Characteristic findings include primarily perivascular and periadnexal localization of the dermal infiltrates, with variable infiltration of the follicular epithelium by small, medium-sized or sometimes large T cells with hyperchromatic and cerebriform nuclei, and sparing of the epidermis (folliculotropism instead of epidermotropism) ( Fig. 120.6B ). Most cases show mucinous degeneration of the follicular epithelium (follicular mucinosis), as assessed with Alcian blue or colloidal iron staining, and some show involvement of the eccrine glands and coils (syringotropism). There is often a considerable admixture with eosinophils and sometimes plasma cells. In the perifollicular infiltrates, the neoplastic T cells may be blast cells rather than cerebriform cells, and therefore may be easily mistaken for histiocytes. In most cases, the neoplastic T cells have a CD3 + , CD4 + , CD8 − phenotype as in classic MF. Admixture with CD30-positive blast cells is common.
Differential diagnosis
The distinctive clinical and histologic features should facilitate an early and correct diagnosis. However, because of the preferential involvement of the head and neck area, the absence of patches and plaques on the trunk and buttocks, and particularly because of the absence of epidermotropic atypical T cells, the diagnosis of MF or CTCL is often not considered and is misinterpreted as seborrheic dermatitis or atopic dermatitis. Clinicopathologic correlation is also required to differentiate folliculotropic MF from other types of CTCL. The relationship between folliculotropic MF and the so-called benign or idiopathic form of follicular mucinosis (alopecia mucinosa) resembles the relationship between classic MF and “parapsoriasis”. In the case of persistent generalized lesions of the idiopathic form, patients should be monitored regularly, since progression into overt folliculotropic MF has been reported.
Treatment
Because of the perifollicular localization of the dermal infiltrates, folliculotropic MF is often less responsive to skin-directed therapies, such as PUVA and topical mechlorethamine, than is classic plaque stage MF. In the case of unresponsive disease, a combination of PUVA with either IFN-α or retinoids, local radiotherapy or TSEB can be used, although sustained complete remissions are rarely achieved .
Pagetoid Reticulosis
▪ Woringer–Kolopp disease ▪ Unilesional MF
Definition
This is a rare variant of MF, characterized by the presence of localized patches or plaques with an intraepidermal proliferation of neoplastic T cells. The term pagetoid reticulosis should only be used for the localized type (Woringer–Kolopp type) and not for the disseminated type (Ketron–Goodman type). Nowadays, patients with generalized disease would most likely be classified as having primary cutaneous CD8-positive aggressive epidermotropic cytotoxic T-cell lymphoma, primary cutaneous gamma/delta T-cell lymphoma, or tumor stage MF .
Epidemiology
Pagetoid reticulosis is extremely rare, and it accounts for fewer than 1% of CTCL cases. It mostly affects adults.
Clinical features
Patients present with a solitary psoriasiform or hyperkeratotic patch or plaque, which is usually localized to an extremity and slowly progressive ( Fig. 120.7A ). In contrast to classic MF, extracutaneous dissemination or disease-related deaths have not been reported.
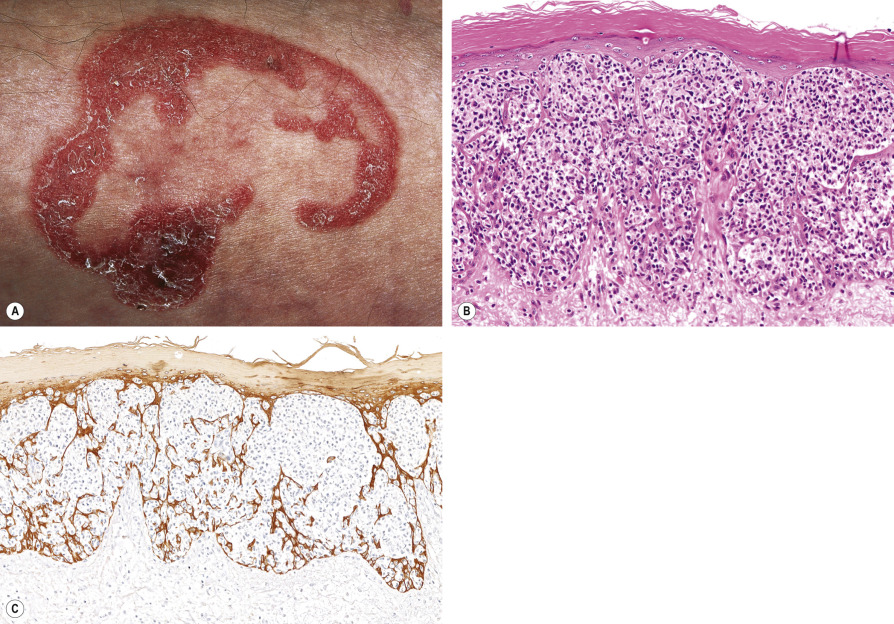
Pathology
The typical histologic picture consists of a hyperplastic epidermis with marked infiltration by large atypical pagetoid cells, arranged singly or in nests or clusters ( Fig. 120.7B ). The atypical cells have medium-sized or large, sometimes hyperchromatic and cerebriform nuclei and abundant, vacuolated cytoplasm. The superficial dermis may have an infiltrate of mostly small lymphocytes, but rarely contains neoplastic T cells.
Immunophenotype
The neoplastic T cells may show either a CD3 + , CD4 + , CD8 − or a CD3 + , CD4 − , CD8 + phenotype. CD30 is often expressed .
Differential diagnosis
Pagetoid reticulosis should be differentiated from other types of epidermotropic CTCL, such as MF, LyP type B, LyP type D, and primary cutaneous CD8-positive aggressive epidermotropic cytotoxic T-cell lymphoma (see Table 120.2 ). Useful criteria for pagetoid reticulosis include the characteristic clinical presentation and the often strictly epidermal localization of the neoplastic T cells.
Treatment
The preferred mode of treatment is radiotherapy or surgical excision.
Granulomatous Slack Skin
Definition
This is an extraordinarily rare type of CTCL characterized by the slow development of folds of lax skin and a granulomatous infiltrate with clonal T cells .
Epidemiology
It is an extremely rare type of CTCL, with ~60 cases reported. It typically affects adolescents and adults, and mostly occurs in men.
Clinical features
This condition is characterized by circumscribed areas of pendulous lax skin with a predilection for the axillae and groin ( Fig. 120.8A ). In approximately one-third of the reported patients, an association with Hodgkin lymphoma was observed. Most patients have an indolent clinical course.
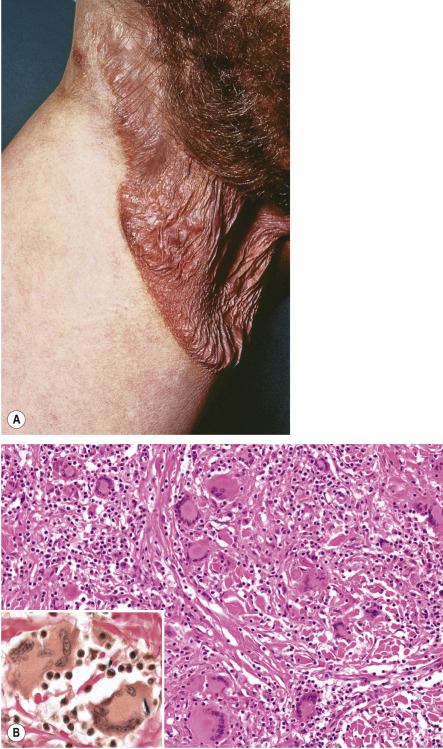
Pathology
Fully developed lesions show dense granulomatous dermal infiltrates containing atypical T cells with cerebriform nuclei, macrophages and often many multinucleated giant cells, as well as destruction of elastic tissue and elastophagocytosis by the multinucleated cells ( Fig. 120.8B ). The epidermis may be infiltrated by small atypical T cells with cerebriform nuclei, as in classic MF. The atypical T cells have a CD3 + , CD4 + , CD8 − phenotype. A large study noted overlapping histologic features between granulomatous slack skin and granulomatous MF .
Treatment
Radiotherapy may be effective, but experience is still limited. Rapid recurrences after surgical excision have been reported.
Sézary Syndrome
Definition
Sézary syndrome (SS) is defined historically by the triad of erythroderma, generalized lymphadenopathy, and the presence of neoplastic T cells (Sézary cells) in the skin, lymph nodes, and peripheral blood (see Table 120.4 ). However, distinguishing SS from benign forms of erythroderma can be extremely difficult. Criteria for the diagnosis of SS include demonstration of a T-cell clone in the peripheral blood (preferably the same T-cell clone as in the skin), in combination with an absolute Sézary cell count ≥1000 cells/mcl or immunophenotypical abnormalities (an expanded CD4 + T-cell population resulting in a CD4/CD8 ratio >10 and/or aberrant expression of pan-T-cell antigens) . While SS is often designated as a leukemic phase or variant of MF, more recent studies have revealed major genomic and phenotypical differences between these two entities, suggesting that SS and MF should be considered as separate lymphomas arising from distinct functional T-cell subsets .
Epidemiology
SS is a rare disease accounting for less than 5% of all CTCL (see Table 120.1 ). It occurs exclusively in adults.
Clinical features
SS is characterized by erythroderma, which may be associated with marked exfoliation, edema and lichenification; it is intensely pruritic ( Fig. 120.9A ). Lymphadenopathy, alopecia, onychodystrophy, and palmoplantar hyperkeratosis are common findings. The overt clinical picture may be preceded by a non-diagnostic dermatitis. The prognosis is generally poor, with an overall 5-year survival of ~25%. Most patients die of opportunistic infections due to immunosuppression.
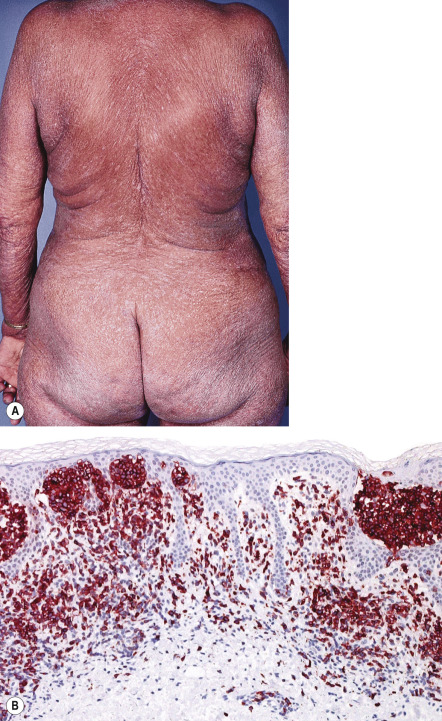
Pathology
The histologic features in SS may be similar to those in MF. However, the cellular infiltrates in SS are more often monotonous, and epidermotropism may sometimes be absent. In up to one-third of skin biopsies from patients with otherwise classic SS, the histologic findings may be nonspecific . Involved lymph nodes characteristically show a dense, monotonous infiltrate of Sézary cells with effacement of the normal lymph node architecture. The bone marrow may be involved, but the infiltrates are often sparse and mainly interstitial. The neoplastic T cells have a CD3 + , CD4 + , CD8 − phenotype, characteristically lack CD7 and CD26 expression, and express programmed death-1 (PD-1; CD279) in almost all cases ( Fig. 120.9B ) .
Pathogenesis
The pathogenesis of SS is unknown. Conclusive evidence of an etiologic role for HTLV-1 is lacking. In SS, the neoplastic CD4 + T cells have the phenotype of a central memory T-cell subset and thus are capable of circulating between skin, lymph nodes and peripheral blood, while the neoplastic cells in MF are derived from non-circulating skin resident effector memory T cells , . In addition to these phenotypical differences, SS and MF have major genomic differences , in particular gross chromosomal instability with highly recurrent gains and losses in SS. These include deletions involving chromosomes 10q22–25 (harbors PTEN ) and 17p12–13 (includes TP53 ) and gains involving chromosomes 8q22–24 (includes MYC ), 10p11.2, and 17q22–25 . Gene expression studies showed increased expression of PLS3 (T-plastin), TWIST1 , and CD158k/KIR3DL2, which might be used as diagnostic markers .
Differential diagnosis
Differentiation between SS and non-neoplastic forms of erythroderma may be extremely difficult . The differential diagnosis includes erythroderma secondary to psoriasis, atopic dermatitis or other forms of dermatitis, pityriasis rubra pilaris, and drug reactions as well as idiopathic erythroderma (see Ch. 10 ). Demonstration of an identical T-cell clone in skin and peripheral blood is an important diagnostic criterion favoring SS. Demonstration of a predominant CD3 + , CD4 − , CD8 + T-cell population in the skin and peripheral blood is highly suggestive of actinic reticuloid (see Ch. 87 ) .
Treatment
Being a systemic disease (leukemia) by definition, systemic treatment is required. Skin-directed therapies like PUVA or potent topical corticosteroids may be used as adjuvant therapy. Extracorporeal photopheresis (ECP), either alone or in combination with other treatment modalities, has been suggested as the treatment of choice in SS and erythrodermic MF, with overall response rates of 30–80%, and complete response rates of 14–25% . This great variation in response rates may reflect differences in patient selection and/or concurrent therapies. The suggested superiority of ECP over traditional low-dose chemotherapy regimens has not yet been substantiated by randomized controlled trials . Beneficial effects have also been reported with IFN-α (either alone or in combination with PUVA therapy) as well as prolonged treatment with methotrexate or low-dose chlorambucil plus prednisone, but complete responses are uncommon. CHOP or CHOP-like regimens may produce higher response rates, but these responses are generally short-lived. Recent studies have observed possible benefits with bexarotene, denileukin diftitox, HDACi and (low-dose) alemtuzumab (anti-CD52), but the long-term effects of these therapies remain to be established . Allogeneic hematopoietic stem cell transplantation may have curative potential, even in patients with advanced disease (see MF treatment).
Introduction
The term cutaneous T-cell lymphoma (CTCL) describes a heterogeneous group of neoplasms of skin-homing T cells that show considerable variation in clinical presentation, histologic appearance, immunophenotype, and prognosis. CTCLs represent approximately 75–80% of all primary cutaneous lymphomas, whereas primary cutaneous B-cell lymphomas (CBCLs) account for approximately 20–25% . For many years, mycosis fungoides (MF) and Sézary syndrome (SS) were the only known types of CTCL. Over the past three decades, based on a combination of clinical, histologic and immunophenotypical criteria, new types of CTCL and CBCL have been defined and new classifications for the group of primary cutaneous lymphomas have been formulated . A major advantage in the management of primary cutaneous lymphomas compared with lymphomas arising at other sites is that the former can be seen and can be biopsied easily, giving the dermatologist the unique opportunity to correlate the clinical appearance and clinical behavior with histologic, immunophenotypical, and genetic aspects of these conditions. Hence, the dermatologist can play a key role in the diagnosis, classification, and treatment of these diseases.
History
In 1806, a French physician, Jean Louis Alibert, was the first to describe a patient with MF. This case was designated pian fungoïde in his atlas, but in 1835 it was renamed mycosis fungoïde because of the resemblance of some skin tumors to mushrooms. In 1870, Bazin described the natural progression from a nonspecific premycotic phase to plaque lesions and finally to tumors, which probably represents one of the first descriptions of the “multistep model” in the development of a malignancy. In 1885, Vidal and Brocq used the term “mycosis fungoides d’emblée” for patients presenting with skin tumors not preceded by patches or plaques. It has now become clear that such cases represent another type of CTCL or a CBCL. The erythrodermic form of MF was described in 1892 by Besnier and Hallopeau. These early descriptions of the major clinical forms of MF were followed by descriptions of Sézary syndrome by Sézary and Bouvrain in 1938, pagetoid reticulosis by Woringer and Kolopp in 1939, and lymphomatoid papulosis (LyP) by Macaulay in 1968.
Thus, in the early 1970s, MF and some related conditions were the only types of cutaneous lymphoma that had been rather well described. Reports on cutaneous lymphomas other than MF/SS, commonly designated in the past as malignant reticulosis or reticulum cell sarcoma, were few. Moreover, they were firmly believed to represent skin manifestations of a systemic lymphoma and treated as such.
The CTCL concept
In 1968, Lutzner and Jordan described the ultrastructural features of the circulating atypical cells in SS. Characteristically, the nuclei of these cells had deep and narrow indentations, giving them a cerebriform appearance. Three years later, similar cells were found in the skin and lymph nodes of patients with MF. In 1975, based on the observation that the neoplastic cells in MF, SS, and related conditions not only had the same morphology but also a common T-cell phenotype, the term “CTCL” was suggested for this group of diseases . Within a short time, this term gained wide acceptance, particularly in the US. The introduction of the CTCL concept can be considered as a landmark in the history of this group of diseases. However, a major disadvantage has been that in many subsequent studies, distinction was no longer made between MF, SS, and other T-cell neoplasms, entities which may vary considerably in clinical presentation and clinical behavior.
The concept of primary cutaneous lymphomas
At about the same time that the CTCL concept was introduced, several European groups started to classify cutaneous lymphomas according to the criteria of the Kiel classification, a system used by hematopathologists for the categorization of nodal lymphomas. It was then determined that many types of CBCL and CTCL (other than classic MF and SS) can present in the skin without any evidence of extracutaneous disease at the time of diagnosis. It appeared that these primary cutaneous lymphomas often have a completely different clinical behavior and prognosis when compared to morphologically similar lymphomas arising within lymph nodes, and therefore require different types of treatment . In addition, differences in the presence of specific chromosomal translocations and in the expression of oncogenes, viral sequences or antigens (e.g. EBV), and adhesion receptors involved in tissue-related lymphocyte homing were found.
Such differences underscored that primary cutaneous lymphomas represent a distinct group and may explain, at least in part, their different clinical behavior. For instance, the observation that the neoplastic T cells in most CTCLs express cutaneous lymphocyte antigen (CLA) and the CC-chemokine receptors 4 (CCR4) and 10 (CCR10) indicates that they are the neoplastic counterparts of normal skin-homing T cells, and this explains why these CTCLs present in the skin. Perhaps most importantly, it appeared that different types of CTCL and CBCL with different clinical behaviors and different therapeutic requirements may have an identical histologic appearance. This implies that histologic features should always be combined with clinical and immunophenotypical data, before a definite diagnosis (classification) is made.
Over the past two decades, such an approach resulted in the delineation of several new types of CTCL and CBCL, and it formed the basis of the European Organization for Research and Treatment of Cancer (EORTC) classification for primary cutaneous lymphomas .
EORTC, WHO and WHO-EORTC classification schemes
The EORTC classification scheme was the first one that was specifically designed for the group of primary cutaneous lymphomas. It contained a limited number of well-defined types of CTCL and CBCL, which together accounted for >95% of all primary cutaneous lymphomas, in addition to some provisional entities, which were not yet fully defined clinically. A distinction was made between cutaneous lymphomas with indolent, intermediate, or aggressive clinical behavior. By including well-defined and recognizable disease entities, this classification provided the clinician with detailed information on staging, preferred mode of treatment, clinical behavior and prognosis, serving as a useful guide to optimal management and treatment.
The third edition of the World Health Organization (WHO) classification for malignant lymphomas adopted the most common types of CTCL from the EORTC classification, but there was controversy with regard to the categorization of some rare types of CTCL . In addition, the WHO classification contained new entities not yet included in the EORTC scheme. In 2004, representatives from both organizations reached an agreement on a new classification scheme for the group of cutaneous lymphomas: the WHO-EORTC classification . Importantly, the fourth edition of the WHO classification for malignant lymphomas, published in 2008 (updated in 2016) and used by pathologists and hematologists worldwide, has adopted the terminology and definitions of the WHO-EORTC classification; therefore, it now includes all the types of CTCL and CBCL included in previous classifications of primary cutaneous lymphomas . The frequency and survival of patients with the different types of CTCL recognized in the WHO-EORTC classification are presented in Table 120.1 . Following a description of practical guidelines for diagnosis, classification and staging, relevant features of the different types of CTCL included in this classification are presented.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


