Abstract
Microvascular occlusion in the skin may have distinctively patterned lesions, such as retiform purpura, and is clinically important to recognize and distinguish from cutaneous vasculitis in order to determine associated disease risk and direct appropriate therapy. This chapter reviews the causes of cutaneous microvascular occlusion, including antiphospholipid antibody syndrome, cryoglobulinemia, heparin-induced thrombocytopenia syndrome, warfarin necrosis, purpura fulminans, and essential thrombocytosis. The cutaneous manifestions of both cholesterol and septic emboli are discussed as are the skin findings of thrombotic microangiopathy, Sneddon syndrome, malignant atrophic papulosis, and livedoid vasculopathy. A helpful algorithm for the clinical approach to a patient with retiform purpura is provided.
Keywords
microvascular occlusion, primary thrombotic microangiopathy, retiform purpura, atrophie blanche, livedoid vasculopathy, malignant atrophic papulosis, cryoglobulins, cryoglobulinemia, cholesterol emboli, heparin-induced thrombocytopenia (HIT), heparin-induced thrombocytopenia syndrome (HITS), myeloproliferative neoplasms, essential thrombocytosis, septic emboli, thrombotic thrombocytopenic purpura, Sneddon syndrome, heparin necrosis, paroxysmal nocturnal hemoglobinuria, warfarin necrosis, warfarin-associated venous limb gangrene, purpura fulminans, antiphospholipid antibody syndrome
Introduction
The differential diagnosis of microvascular syndromes involving the skin is extensive and it is critical to distinguish between inflammatory injury and non-inflammatory occlusive injury to vessels. The diagnosis of occlusive syndromes in the skin is greatly facilitated by recognizing the telltale lesions of retiform purpura or non-inflammatory (bland) necrosis.
Diseases that produce skin lesions secondary to microvascular occlusion have often been lumped with cutaneous vasculitic syndromes, or discussed primarily as systemic diseases with little attention paid to cutaneous findings. While there is some overlap in the pathogenesis of deep venous thrombosis and pulmonary emboli and the pathogenesis of cutaneous microvascular occlusion syndromes, the two differ substantially with respect to disease etiology and treatment.
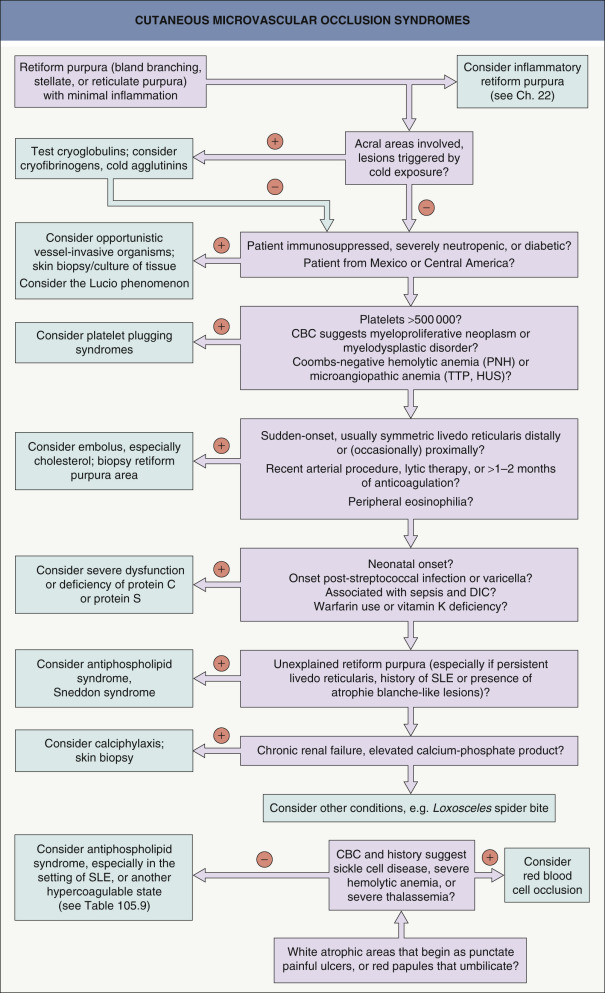
The differential diagnosis for occlusive syndromes is best approached through major pathophysiologic categories ( Table 23.1 ). These categories frequently have sufficiently distinctive clinical settings that a minimal history and physical examination plus laboratory findings quickly allows for an efficient sorting of diagnostic possibilities ( Fig. 23.1 ). This is especially important in life-threatening situations where making timely decisions is crucial. A reasonable panel of initial laboratory tests is listed in Table 23.2 .
| DIFFERENTIAL DIAGNOSIS OF CUTANEOUS MICROVASCULAR OCCLUSION BASED ON PATHOPHYSIOLOGY | |
| Platelet-related thrombopathy | |
| |
| Cold-related precipitation or agglutination | |
| |
| Vessel-invasive organisms | |
| |
| Embolization | |
| |
| Systemic coagulopathies | |
| Vascular coagulopathies | |
| |
| Cell occlusion syndromes | |
| |
| Miscellaneous/unknown mechanism | |
|
|
| BASIC SCREENING TESTS FOR OCCLUSIVE SYNDROMES |
| Complete blood count with differential, platelet count, and blood smear |
|
| Partial thromboplastin time (PTT) |
|
| Cryoglobulins (occasionally cryofibrinogen and cold agglutinins) when location and history suggest cold occlusion syndrome |
| Biopsy |
|
| Basic hepatic and renal function screens |
|
| ANCA |
|
It must be emphasized that some inflammatory syndromes capable of producing retiform purpura may occasionally present with lesions that have minimal inflammation (see Ch. 22 ). In particular, this may occur with the antineutrophil cytoplasmic antibody (ANCA)-positive syndromes of granulomatosis with polyangiitis (GPA) and microscopic polyangiitis (MPA), as well as ANCA-negative cutaneous polyarteritis nodosa.
Because treatment depends on pathogenesis (see Fig. 22.3 ), accurate diagnosis is critical prior to instituting appropriate therapy. The use of anti-inflammatory therapy for occlusive disease may not only be useless but actually harmful, and the same may be true for anticoagulant therapy in most vasculitides.
Disorders of Platelet-Related Thrombopathy
Heparin-Induced Thrombocytopenia Syndrome
▪ Heparin-induced thrombocytopenia (HIT) syndrome – platelet activation stage ▪ Heparin-associated thrombocytopenia with thrombosis (HATT) syndrome – includes patients who develop thrombosis
- ▪
Exposure to heparin, most often unfractionated
- ▪
Retiform, branching purpura or non-inflammatory necrosis
- ▪
Lesions may arise locally at sites of heparin infusion/injection or at distant sites
- ▪
Detection of anti-platelet factor 4-heparin complex antibodies via ELISA has a very high negative predictive value but low positive predictive value
Introduction
Heparin necrosis is an unusual but important iatrogenic syndrome first recognized in the early 1970s. Necrosis may occur after subcutaneous or intravenous heparin administration, including the trace amounts of heparin used to keep infusion lines open. Of note, the same pathogenic cascade may be activated even without heparin exposure (see below) .
Heparin-induced thrombocytopenia syndrome (HITS) occurs in 1–5% of adults exposed to heparin, with 30–90% of these patients developing thrombosis. However, the latter figures are primarily derived from retrospective series in which the diagnosis was made following a thrombotic episode; as a result, it is likely that the true incidence of thrombosis is 30–50%.
Epidemiology
HITS occurs in ~1 in 5000 hospitalized patients . The risk of developing HITS is ten times greater in patients receiving unfractionated heparin, as compared with low-molecular-weight heparin. Risk is also higher following major surgery, particularly cardiac bypass. Although slightly more common in women compared to men, it is rare during pregnancy.
Pathogenesis
In Table 23.3 , the physiologic pathway in which binding of platelet factor 4 (PF4) to polyanions on the surface of bacteria leads to their phagocytosis is compared to the pathologic pathway that underlies HITS. The latter involves the binding of PF4 to the polyanion heparin .
| HEPARIN-INDUCED THROMBOCYTOPENIA SYNDROME (HITS) – ROLE OF PLATELET FACTOR 4 |
| Platelet factor 4 (PF4) – physiologic pathway |
|
| Platelet factor 4 (PF4) – pathologic pathway in HITS |
|
Clinical features
HITS usually occurs between days 5 and 10 of heparin therapy . In those patients exposed to heparin within the past 90–100 days, HITS may develop within hours after re-exposure. Occasionally, these individuals experience an anaphylactoid reaction within 30 minutes of a heparin bolus . There is also a delayed-onset form of heparin necrosis, in which lesions can occur up to 3 weeks after exposure to heparin. Of note, heparin-dependent antibodies do not invariably reappear with subsequent heparin use after 100 days.
There are additional unusual clinical scenarios in which HITS can also develop. In patients receiving chronic heparin, e.g. for renal dialysis, major surgery may trigger HITS within 5–10 days. Even in the absence of exposure to heparin, major surgery or bacterial infections can trigger a spontaneous or autoimmune HIT syndrome. Presumably the latter is due to PF4 binding to DNA, RNA, or glycosaminoglycans and induction of the PF4 pathologic pathway (see Table 23.3 ).
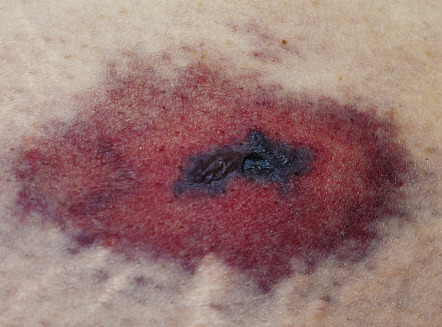
Lesions are usually tender, sharply demarcated, non-inflammatory, and purpuric or necrotic, often with a retiform morphology. Large necrotic areas with or without branching margins can also be seen. Lesions may occur at the site of subcutaneous injections ( Fig. 23.2 ) or may develop at sites distant to the heparin infusion (see Fig. 21.22 ) . Although consumption of platelets is part of this syndrome, absolute thrombocytopenia is often not present in patients with significant vascular occlusion. A decline in the platelet count of >50% from the most recent highest value prior to the administration of heparin (preferably from the day heparin is begun) should prompt consideration of HITS. Any organ or limb is potentially at risk for vascular occlusion, including the CNS.
Pathology
If HITS is associated with thrombosis, there is non-inflammatory occlusion of blood vessels and signs of ischemic necrosis. Microvascular as well as venous or arterial occlusion can be seen, both in the skin and internal organs.
Differential diagnosis
A recent history of heparin administration should prompt consideration of this diagnosis in any patient who develops retiform purpura or bland necrosis, whether or not the patient is thrombocytopenic. Of note, a decrease in the platelet count is a common incidental finding in patients who are ill and coincidently receiving heparin and therefore can be unrelated to the development of this syndrome. Conversely, a normal platelet count does not exclude the diagnosis. Other cutaneous occlusion syndromes associated with thrombocytopenia include antiphospholipid antibody syndrome, purpura fulminans secondary to sepsis, and thrombotic thrombocytopenic purpura (although skin lesions due to microvascular occlusion are rare in the latter syndrome). While specialized testing can confirm the presence of anti-PF4–heparin complex antibodies, a clinical diagnosis is often needed before the results of this test are known. In addition, detection of anti-PF4–heparin complex antibodies via ELISA has a 98–99% negative predictive value but low positive predictive value .
Treatment
Treatment requires cessation of heparin therapy and initiation of therapeutic anticoagulation. Argatroban, a direct thrombin inhibitor, is FDA-approved for the treatment of HITS and outside of the US, danaparoid, a factor Xa inhibitor, is an approved therapy (see Table 22.8 ) . Although not FDA-approved for this indication, fondaparinux and bivalirudin have also been found to be effective. Whereas patients with HITS and thrombosis require therapeutic anticoagulation for at least three months, treatment duration for patients with only HITS has not been standardized. Importantly, vitamin K antagonists (e.g. warfarin, phenprocoumon) are contraindicated during the acute phase because of an increased risk of developing venous limb gangrene secondary to decreased protein C levels . Patients with delayed or spontaneous HITS should be anticoagulated with non-heparin agents (e.g. argatroban, bivalirudin). Plasma exchange has been used for refractory HITS .
Myeloproliferative Neoplasms
Essential thrombocytosis: primary thrombocythemia
- ▪
Myeloproliferative neoplasms include essential thrombocytosis, polycythemia vera, primary myelofibrosis, and chronic myelogenous leukemia
- ▪
In the first three disorders, mutations have been detected in JAK2 , MPL , and CALR and these genes encode a tyrosine kinase, the thrombopoietin receptor, and calreticulin, respectively; in polycythemia vera, ~95% of patients have activating JAK2 mutations
- ▪
Depending upon the degree of platelet count elevation, thrombosis and/or bleeding can occur
Introduction
In patients with myeloproliferative neoplasms, thrombocytosis is a risk factor for vascular occlusion. As a result, individuals with essential thrombocytosis are most likely to develop cutaneous lesions due to microvascular occlusion, followed by those with polycythemia vera. Some patients with myeloproliferative neoplasms can eventually develop myelodysplastic syndrome or acute myelogenous leukemia.
Pathogenesis
Myeloproliferative neoplasms are clonal hematopoietic disorders whose manifestations depend on the particular type of stem cell affected. A single mutation, JAK2V617F, is present in 90–95% of polycythemia vera patients and 50–70% of those with essential thrombocytosis and primary myelofibrosis. The JAK2 mutations lead to activation of the JAK–STAT pathway (see Fig. 128.10 ). In the remainder of patients, mutations in MPL and CALR occur more commonly in essential thrombocytosis and primary myelofibrosis than in polycythemia vera where they are rare .
In myeloproliferative and myelodysplastic disorders, platelets can function abnormally, leading to occlusion and/or hemorrhage. When the platelet count is 400–1000 × 10 9 /L, thromboembolism is common, whereas both thrombosis and bleeding may occur with a platelet count of 1000–2000 × 10 9 /L; bleeding predominates when the platelet count is >2000 × 10 9 /L . Acquired von Willebrand syndrome occurs when the platelet count is >1000 × 10 9 /L, providing an explanation for the associated hemorrhage. When the platelet count is within the normal range in the setting of a myeloproliferative neoplasm, the risk of thrombosis is uncertain.
Epidemiology
Both polycythemia vera and essential thrombocytosis occurs more often in women. They are uncommon before the age of 50 years, but the incidence increases almost exponentially after age 60 years. Patients with polycythemia vera have an increased cardiovascular mortality risk (1.5 deaths/100/year) and a non-fatal thrombosis risk of 3.8 events/100/year, with equal occurrence of arterial and venous events . Individuals with essential thrombocytosis have a risk of 2–4% per patient-year of fatal and non-fatal events, with an arterial : venous ratio of 2–3 : 1.
Clinical features
Cutaneous lesions occur in ~20% of patients with essential thrombocytosis, including purpura, hematomas, livedo reticularis, erythromelalgia, Raynaud phenomenon, urticaria, cutaneous small vessel vasculitis, leg ulcers, gangrene, and recurrent superficial thrombophlebitis . An important subset of these cutaneous lesions is due to microvascular occlusion.
Erythromelalgia is a syndrome that can occur as a primary (idiopathic) or a secondary phenomenon (see Ch. 106 ). It is characterized by intense burning and paroxysmal bright erythema of the distal extremities. Most closely associated with essential thrombocytosis, and less often with polycythemia vera, erythromelalgia can occur with any myeloproliferative or myelodysplastic disorder if the platelet count is sufficiently elevated . Because this form of secondary erythromelalgia is caused by platelet-mediated acral vasodilation, inflammation and microvascular occlusion, patients can also develop discrete areas of purpura or necrosis, often retiform in outline, as a unique manifestation. This version of erythromelalgia typically responds dramatically to aspirin administration, whereas idiopathic and other secondary forms generally show minimal or no benefit from aspirin.
Other clinical features of patients with myeloproliferative neoplasms depend on the diagnosis, e.g. polycythemia patients may have a ruddy cyanosis and experience severe pruritus following exposure to warm water . Chronic myelogenous leukemia patients have elevated neutrophil counts and, frequently, elevated eosinophil or basophil counts.
Pathology
Microvascular occlusion of dermal vessels or subcutaneous arterioles is found in biopsy specimens from retiform purpura or necrotic lesions.
Differential diagnosis
Useful findings which suggest a myeloproliferative neoplasm include anemia or elevated hematocrit, an abnormal white blood cell count or differential, and an abnormal platelet count or platelet morphology. The differential diagnosis of reactive thrombocytosis, especially due to iron deficiency, infection or malignancy, must be considered when assessing the significance of an elevated platelet count.
Treatment
Low-risk patients with polycythemia vera (PV) and essential thrombocytosis (ET) may be treated with low-dose aspirin. First-line therapy for high-risk patients (>65 years [PV] or >60 years [ET] or history of thrombosis) is low-dose aspirin plus cytoreductive therapy, typically hydroxyurea or interferon-α .
Prompt and lasting relief of burning pain by low-dose aspirin is a characteristic finding in thrombocythemic erythromelalgia. The refractory pruritus in polycythemia vera may respond to paroxetine (20 mg/day), interferon-α, UVB phototherapy, or ruxolitinib (JAK inhibitor) . Imetelstat, a telomerase inhibitor, is FDA-approved for myelofibrosis and is currently under investigation for the treatment of essential thrombocytosis .
Paroxysmal Nocturnal Hemoglobinuria
- ▪
Coombs-negative intravascular hemolytic anemia due to mutations in PIG-A
- ▪
Also cytopenias and thrombosis
- ▪
Cutaneous lesions are uncommon, but may be extensive
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare hematopoietic stem cell disorder that is associated with an increased risk of thrombosis .
Pathogenesis
This disease is due to a somatic mutation in PIG-A ( p hosphatidyl i nositol g lycan class A ) in hematopoietic stem cells. This X-linked gene encodes a glycosylphosphatidylinositol (GPI)-anchored protein that is required for the cell membrane attachment of several important membrane proteins. These proteins are responsible for protecting blood cells and platelets from complement-mediated injury (e.g. CD55, CD59). The relative lack of protection leads to red cell lysis and activation of platelets.
The cause of venous thrombosis is multifactorial and is due in part to microparticle formation from platelets and other blood component membranes as well as depletion of nitric oxide related to hemolytic release of free hemoglobin.
Clinical features
This acquired clonal hematologic disorder classically presents with intravascular hemolysis, deficient hematopoiesis, and a thrombotic tendency. It may also occur in the setting of hematologic disorders such as aplastic anemia and myelodysplastic syndrome where it is often subclinical because of a limited number of mutated clones . Thrombotic events are the most common cause of death , and venous thromboses (e.g. Budd–Chiari syndrome) are more common than arterial. A variety of cutaneous findings have been reported in PNH, including hemorrhagic bullae, petechiae, leg ulcers, non-inflammatory retiform purpura, and a purpura fulminans-like presentation .
Depletion of nitric oxide related to chronic hemolysis can lead to smooth muscle dystonia with dysphagia, abdominal pain, and erectile dysfunction . There is also a six-fold increased risk of chronic kidney disease.
Pathology
Biopsy specimens reveal microvascular occlusion. Detection of an absence or severe deficiency of GPI-anchored protein in ≥2 cell lineages (e.g. WBCs, RBCs) by flow cytometry of peripheral blood is diagnostic of PNH .
Differential diagnosis
The differential diagnosis of the various skin lesions that can occur in PNH is broad. The presence of hemolytic anemia plus pancytopenia should suggest the diagnosis.
Treatment
In the past, treatment was largely supportive, consisting of transfusions, iron and folic acid supplementation, and anticoagulation when needed. Eculizumab, which inhibits the terminal phase of the complement cascade at the C5 stage and can reduce intravascular hemolysis and venous thrombosis, is now routinely prescribed as a lifelong therapy . Because eculizumab can increase the risk of meningococcemia via C5 inhibition, patients should ideally receive the meningococcal vaccine at least 2 weeks before instituting therapy.
Primary Thrombotic Microangiopathy
- ▪
Thrombotic microangiopathy (TMA) is characterized by microangiopathic hemolytic anemia (RBC fragmentation) and thrombocytopenia
- ▪
While previously categorized as either thrombotic thrombocytopenic purpura (TTP) or hemolytic uremic syndrome (HUS), there are multiple subtypes of primary TMA and causes of secondary TMA
- ▪
Round, macular petechiae are seen in inherited or acquired ADAMTS13-related TMA [TTP], but skin lesions are not observed in Shiga toxin-mediated TMA [HUS]
Introduction
Multiple subtypes of thrombotic microangiopathy (TMA) have been identified that can mimic classic TTP or HUS ( Table 23.4 ) .
| SUBTYPES OF PRIMARY AND DRUG-INDUCED THROMBOTIC MICROANGIOPATHY (TMA) | |||||
| Subtype of TMA | Etiology | Pathomechanism | Onset | Clinical features | Treatment |
| Inherited | |||||
| ADAMTS13 deficiency-mediated [thrombotic thrombocytopenic purpura] | Homozygous or heterozygous mutations in ADAMTS13 | ADAMTS13 is a vWf-cleaving protease and its dysfunction leads to vascular occlusion from ultra-large vWF multimers | Childhood, occasionally adulthood | Recurrent TMA; neurologic or other organ (but rarely severe renal) injury Petechiae | Plasma infusion, plasma exchange |
| Complement-mediated (90% of cases inherited) | Mutations in genes that encode components of the alternate complement pathway, e.g. H, I, B, C3, CD46 | Uncontrolled activation of the alternative complement pathway | Often childhood, sometimes adulthood | TMA; renal injury common | Anti-complement therapy (e.g. eculizumab), plasma exchange or infusion |
| Metabolism-mediated | Mutations in MMACHC lead to AR disorder methylmalonic aciduria and homocystinuria, CbIC type | Metabolic abnormalities trigger platelet activation, formation of reactive oxygen species, endothelial dysfunction, ↑tissue factor, and coagulation | Infancy (before 1 year of age) | Developmental delay, hypotonia; pulmonary artery hypertension; chronic kidney disease with hypertension and proteinuria | B12, betaine, folinic acid |
| Coagulation-mediated | Mutations in THBD , PLG , and DGKE which encode thrombomodulin, plasminogen, and diacylglycerol kinase epsilon (DGKE), respectively | DGKE dysfunction leads to upregulation of prothrombotic factors and downregulation of VEGF receptor | Usually infancy (before 1 year of age) | Acute kidney injury | Plasma infusion |
| Acquired | |||||
| ADAMTS13 deficiency-mediated [thrombotic thrombocytopenic purpura] | Autoantibodies lead to inhibition of ADAMTS13 activity (to <10% of normal) | Decreased activity of ADAMTS13 leads to vascular occlusion from ultra-large vWF multimers (see above) | Adulthood (usually 20–50 years of age) > childhood More common in women and blacks | TMA with thrombocytopenia; GI symptoms, focal neurologic defects, transient renal injury Petechiae | Plasma exchange (improves survival from 10% to ~80% but must be instituted early) |
| Shiga toxin (ST)-mediated [hemolytic uremic syndrome] | Enteric infection with a toxin-secreting strain of either Escherichia coli (especially O157:H7) or Shigella dysenteriae | Toxin binds to Gb3 (CD77, ceramide trihexoside) on endothelial, renal epithelial, and mesangial cells, with binding leading to apoptosis Toxin also prothrombotic | Early childhood (median age = 2 years; 3% mortality) > adulthood (more severe) | Infectious enterocolitis As diarrhea resolves, thrombocytopenia and renal disease develop | Supportive care including aggressive hydration, dialysis often required |
| Drug-mediated, immune type | Most commonly quinine, quetiapine, gemcitabine; also interferon-β (more likely than toxic) | With quinine, drug-dependent antibodies | Adulthood (drug exposure more likely) | Sudden onset of severe systemic symptoms; anuric renal injury | Discontinuation of drug, supportive care |
| Drug-mediated, toxic/dose-related | Multiple drugs, including cyclosporine, tacrolimus, VEGF inhibitors | Multiple mechanisms | Adulthood (drug exposure more likely) | Gradual onset of renal failure | Discontinuation of drug, supportive care |
| Complement-mediated (10% of cases acquired) | Antibodies inhibit complement factor H activity | Reduced inhibition of alternate complement pathway factors (i.e. enhanced complement activation) leads to vessel damage | Childhood or adulthood | Acute kidney injury | Plasma exchange, immunosuppression, anti-complement therapy (e.g. eculizumab) |
Pathogenesis
Table 23.4 outlines the pathogeneses of various subtypes of TMA.
Clinical features
The complete syndrome of classic TTP is characterized by fever, petechiae, thrombocytopenia, microangiopathic hemolytic anemia (schistocytes or fragmented red cells prominent on peripheral smear), renal disease, and neurologic symptoms (most commonly headache and confusion). The macular petechiae usually reflect simple hemorrhage, but occasionally may be attributed to platelet plugging. While platelet occlusion has been thought to occur primarily in visceral organs but not the skin, very few papers have addressed the histology of cutaneous lesions. Nowadays, the classic syndrome of TTP would be classified as within the spectrum of ADAMTS13 deficiency-mediated TMA, either inherited or acquired (see Table 23.4 ). Of note, other subtypes of primary TMA seldom have associated cutaneous hemorrhage.
Disease may remain subclinical unless triggered by factors such as drugs, pregnancy, autoimmune connective tissue disease, systemic infections, hematopoietic stem cell transplant, or malignant hypertension. This is referred to as secondary TMA.
Pathology
Microvascular occlusion of visceral terminal arterioles and capillaries is expected, but is rare in the cutaneous microvasculature. Petechial (cutaneous) hemorrhage is most likely due to simple hemorrhage, but documentation is limited.
Differential diagnosis
The constellation of fever, CNS findings, renal disease, microangiopathic findings in the blood smear (red cell fragmentation), and thrombocytopenia can be seen in sepsis syndromes with disseminated intravascular coagulation (DIC) and rarely in the antiphospholipid antibody syndrome. A normal or minimally elevated prothrombin time plus profound thrombocytopenia strongly favors primary TMA over DIC in adults . Sepsis with purpura fulminans and DIC is characterized by elevation of the prothrombin time and partial thromboplastin time.
Treatment
Treatment of primary and drug-induced TMA is reviewed in Table 23.4 .
Disorders of Cryoprecipitation or Cryoagglutination
- ▪
Retiform purpura in an acral location
- ▪
Due to cryoglobulins, cryofibrinogen, or rarely cold agglutinins
- ▪
Cold-induced cryoprecipitation and vascular occlusion most commonly associated with type I cryoglobulins
- ▪
In acute lesions, eosinophilic hyaline deposits occlude blood vessels
Introduction
Cryoglobulins are immunoglobulins that are present in both serum and plasma and reversibly precipitate with cold exposure . Cryofibrinogens are fibrinogens which precipitate in the cold, are consumed in clotting, and are therefore detectable only in plasma samples . Cold agglutinins are antibodies which promote agglutination of red cells on exposure to cold. All three groups can cause occlusive syndromes in the skin that are triggered by cold exposure. While cryofibrinogens and cold agglutinins can be detected in many patients in association with a variety of illnesses, they are rarely the cause of cold-related occlusion syndromes . Clinical syndromes due directly to cryoprecipitation or cryoagglutination are uncommon.
Pathogenesis
Cryoglobulins change their configuration in the cold and become water-insoluble, which increases blood viscosity leading to sludging and occlusion. However, many of these proteins transform only at refrigerator temperatures, not at skin temperatures typically seen with cold exposure of acral areas. Cryoglobulins cause disease via two mechanisms: occlusion (type I) or immune complex-mediated vasculitis (types II, III) . Simple occlusion with minimal early inflammation and often retiform purpura/necrosis develops when cryoproteins precipitate upon cold exposure, and this is primarily a reflection of monoclonal immunoglobulins (IgG, IgM ≫ IgA, light chains), i.e. type I cryoglobulinemia that is due to an underlying plasma cell dyscrasia or lymphoproliferative disorder. Immune complex disease leads to inflammatory purpura that is palpable, and this is related to mixed cryoglobulinemia (types II or III), often due to hepatitis C viral infection (see Table 24.6 ).
Cryofibrinogen precipitate is composed of fibrinogen, fibrin, fibronectin, and occasionally albumin, immunoglobulins, or other plasma proteins . Cold agglutinins bind to red cells in the cold, often triggering complement activation and red cell lysis. Because of their ability to bind multiple red cells, pentameric IgM antibodies much more readily agglutinate red cells.
Epidemiology
The prevalence of cryofibrinogenemia varies from 0% to 7% in healthy individuals and from 8% to 13% in hospitalized patients . Its female : male ratio is 1.5–4.5 : 1, with most studies favoring the lower end. Cryofibrinogenemia may be essential or secondary to a wide spectrum of disorders including infections, autoimmune diseases, and carcinomas. When a cryopathy has been suspected, cryofibrinogenemia was observed in 10–50% of patients, although usually not in isolation.
Clinical features
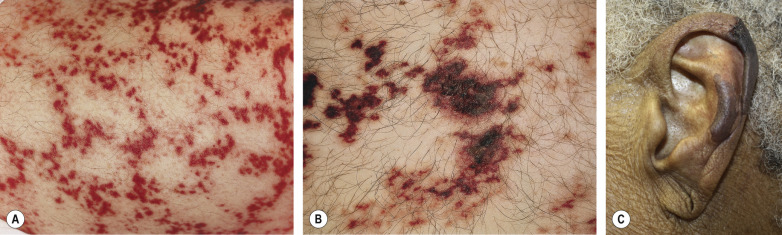
The cardinal sign of cold occlusion, including that due to cryoglobulinemia , is purpuric or necrotic lesions, often retiform, at acral sites of cold exposure ( Fig. 23.3 ). Other cutaneous findings include acral cyanosis, Raynaud phenomenon, and livedo reticularis. Although some reports describe cutaneous small vessel vasculitis in patients with type I cryoglobulins, it is likely that this finding is a secondary leukocytoclastic vasculitis that occurs following occlusive necrosis or ulceration (see Fig. 22.3 ). Mixed (types II and III) cryoglobulins may cause occlusion if they are unstable at close to body temperature, but more frequently they induce lesions via immune complex mechanisms, with clustering of classic palpable purpura in dependent areas.