Cutaneous Lymphoma: Introduction
|
Introduction
Cutaneous lymphomas (CLs) represent clonal proliferations of neoplastic T or B lymphocytes and rarely of natural killer (NK) cells or—for historical reasons— plasmacytoid dendritic cells. CLs have been recognized as a heterogeneous group with distinct variability in clinical presentation, histopathology, immunophenotyping, and prognosis.
Primary CLs often show a completely different clinical behavior and prognosis than do histologically similar systemic lymphomas, which may involve the skin secondarily. Therefore, primary CLs require different approaches to treatment. For this reason, a consensus classification has been created based on both the European Organisation for Research and Treatment of Cancer (EORTC) classification for primary CLs and the World Health Organization (WHO) classification for tumors of hematopoietic and lymphoid tissues. This first common classification (WHO-EORTC)1 categorizes the entities according to lineage and then according to a combination of morphology, immunophenotype, genetic features, and clinical syndromes (Box 145-1) and constituted the basis for the classification of CLs in the new WHO classification 2008.2 This chapter discusses the most frequent cutaneous T-cell lymphomas (CTCLs)—mycosis fungoides (MF), Sézary syndrome (SS), primary cutaneous anaplastic large-cell lymphoma (cALCL), and lymphomatoid papulosis (LyP)—and the most frequent cutaneous B-cell lymphomas (CBCLs)—primary cutaneous follicle center lymphoma (PCFCL), primary cutaneous marginal zone lymphoma (PCMZL), and primary cutaneous diffuse large B-cell lymphoma (PCLBCL), leg type. These seven types of cutaneous lymphoma represent nearly 90% of all CLs.3 Rare entities occurring primarily in the skin are also described.
Cutaneous T-Cell and NK-Cell Lymphomas
Cutaneous B-Cell Lymphomas
|
Epidemiology
Primary Cutaneous T-Cell Lymphomas
CTCLs are non-Hodgkin lymphomas characterized by a dominant skin-homing T-cell clone. They represent approximately 80% of all CLs. The most common forms (approximately 65% of CTCL) are MF and SS, with an annual incidence of five new cases per million people. After MF and SS, the primary cutaneous CD30+ lymphoproliferative disorders comprising LyP and cALCL represent the second most common group of CTCLs (approximately 27%).
Etiology
For many years, CTCLs were thought of as a disease of long-term antigen stimulation in which an initial inflammatory response in the epidermis leads to T-cell proliferation and, finally, to the emergence of a malignant clone.6 Despite much effort to identify such an antigen, the etiology remains unknown.
Due to the above-mentioned hypothesis of antigen stimulation several studies have analyzed the HLA background of affected individuals. Two independent studies showed an association of distinct HLA class II molecules and MF or SS, i.e., the alleles HLA-DRB1*11 and DQB1*03 are significantly overrepresented in these patients.7,8
A viral etiology for CTCLs was an attractive hypothesis given the cutaneous similarities to adult T-cell lymphoma/leukemia, which is associated with human T-cell lymphotrophic virus type 1 (HTLV-1) infection (see Chapter 197). The results of studies to determine an association between CTCL and HTLV-1 are contradictory. Although few studies reported detection of HTLV-1 or -2 sequences by polymerase chain reaction (PCR) analysis in peripheral blood mononuclear cells (PBMCs) or skin biopsies of CTCL patients,9,10 most reports of American and European populations repeatedly failed to demonstrate an association of MF with HTLV-1 infection by PCR, Southern blot, and reverse transcriptase assays.11–13 These data suggest that HTLV-1 does not play an important role in the etiology of CTCLs, and that the only reason to screen patients for antibodies is suspicion that the diagnosis is adult T-cell lymphoma/leukemia rather than MF. Epstein-Barr virus (EBV) has also been proposed as a causative pathogen.14 Given its association with other lymphoproliferative disorders and lymphomas, such as Burkitt lymphoma, this seemed logical. Several studies have shown that EBV is detectable only in a minor percentage of CTCL lesions. In these studies, EBV detection was related to a poor prognosis and its presence is more likely related to immunosuppression caused by either the disease or the therapy, than to etiology of CTCL.15 However, a strong association of EBV and a rare cutaneous lymphoproliferative disease with a hydroa vacciniforme-like appearance, which occurs mostly in people of Asian origin has been observed.16
Bacterial infections have also been implicated in the etiology of CTCLs.17 Of special interest has been the hypothesis that superantigens from Staphylococcus aureus may be responsible for chronic antigenic stimulation.18 In several studies, S. aureus has been detected in a high percentage on the skin of CTCL patients with a high tumor burden, while patients in early stage disease did not show significant differences to control groups.19,20 Though these studies conclusively demonstrated the involvement of S. aureus in disease exacerbation and clinical improvement following antibiotic treatment, the missing difference in S. aureus colonization of early stages of CTCL and control groups questions the involvement of S. aureus or superantigens produced by these bacteria in initiation of CTCLs.
Besides infectious pathogens, it has also been suggested that environmental and occupational risk factors play a causative role in CTCL, because an indolent dermatitis often precedes the diagnosis. Exposure to carcinogens in the work environment could provide the suspected long-term antigenic stimulation for the initiation of the clonal expansion. In epidemiologic studies,21,22 several occupations like glass formers, potters, or paper and wood industry workers have been associated with a higher risk for development of MF. However, neither are the results of the different studies consistent nor could a common denominator like exposure to known carcinogens be identified. With regard to chronic antigenic stimulation by occupational contact allergens it has to be taken into account that MF arises typically on body areas like the lateral trunk, which are protected by clothing during working time. Also other environmental risk factors, like consumption of alcohol, smoking, or exposure to ultraviolet (UV) radiation, were not consistently observed in association with an increased risk for CTCLs.
Pathogenesis
The clinical entities encompassed by the term cutaneous T-cell lymphomas share several components: the epidermal and/or dermal microenvironment, a clonal T-cell population, and a modulated antitumor response.23,24 It has to be kept in mind that due to the rare occurrence of CTCLs in general and of the less prevalent subtypes in particular, nearly all of the knowledge on pathogenetic events has arisen from studies on specimens from MF and SS patients.
The basis for CTCLs as a malignancy confined to the skin, at least in the initial stages of the disease, is the expression of skin-specific homing receptors on the malignant T-cell clone. Skin-homing T cells can be distinguished from other T cells by a unique cell surface receptor called cutaneous lymphocyte-associated antigen (CLA). CLA is an inducible posttranslational carbohydrate modification of the ubiquitously expressed T-cell surface protein, P-selectin glycoprotein ligand 1 by α(1,3)-fucosyltransferase and is expressed on 30% of memory T cells in the peripheral blood.25 It has been shown that induction of CLA expression on T cells occurs during the transition from naive to memory T cells and is mediated by dendritic cells isolated from peripheral skin draining lymph nodes.26 CLA-positive cells have the ability to home to the skin by binding to E-selectin, which is expressed on endothelial cells of dermal postcapillary venules and is superinduced during cutaneous inflammation. As T cells in CTCLs express CLA and CD45RO, a memory T-cell marker, it is though that CTCLs arise from skin homing memory T cells. However, as CLA expression is strongly detectable in skin biopsies from both MF and inflammatory dermatoses, it cannot be used for the distinction of malignant and reactive T cells or malignant from inflammatory skin conditions.27 In addition to CLA, expression of the chemokine receptors CCR4 and CXCR3 has been implicated in homing of the malignant cells to the epidermis and dermis. Furthermore, the respective ligands, i.e., the chemokines CCL17 and CCL22 have been found in skin lesions of CTCLs.28 Interestingly, diminished expression of these skin homing receptors seems to be associated with more aggressive disease, i.e., large cell transformation, tumor formation. and dissemination of tumor cells into the blood or lymph nodes.29,30
Besides chemokines that attract the T cells into the skin, cytokines play an important role in the pathogenesis of CTCLs. Because of the memory phenotype of the tumor cells the cytokines interleukin 7 (IL-7) and IL-15, which are necessary for long-term survival and basal homeostatic proliferation of CD4 memory T cells, are of special interest.31 In vitro studies and immunohistochemical analyses on skin biopsy specimens from lesions of MF demonstrate the presence of IL-15 and IL-7 and their capacity to prolong survival of a cell line derived from a SS patient in vitro, probably by an upregulation of the antiapoptotic protein bcl-2. As source of these cytokines both keratinocytes and the tumor cells themselves have been discussed.32,33
Direct genetic analysis of the clonal T cells and the identification of pathogenetically relevant genes in CTCLs have been hampered by the difficult differentiation of malignant and reactive lymphocytes in skin specimens on the one hand and by a large variety of reported genetic alterations on the other. The basis for the latter phenomenon is a chromosomal instability in tumor cells of patients with CTCLs, which has been described repeatedly.34 In the last years, the application of large-scale genome analysis by comparative genomic hybridization and array technologies has elucidated the genetic basis for established pathogenetic factors and disclosed several new pathways, which are of central importance in the pathogenesis of CTCLs. These studies were conducted on samples with high tumor load from MF (skin biopsy specimens from tumors) and SS (PBMCs) patients, and also demonstrated considerable differences of these two entities in regard to genetic abnormalities.35 The most consistent results were obtained for chromosomal and transcriptional gains affecting the proto oncogene MYC and loss of the tumor suppressor gene TP53, a combination for which it is shown to induce genetic instability by disrupting cell cycle control and apoptosis.36 Furthermore, gains of genes important for the generation of survival signals, for example, the cytokine IL-7, the IL-2 receptor or transcription factors like STAT3/5, as well as alterations in members of the NFκB pathway, have been described.37 In addition to gains, losses of well-known tumor suppressor genes, for example, CDKN2A (p16), were detected.38
Loss of Fas expression on tumor cells is of special importance as the Fas receptor/Fas ligand (FasL) pathway is involved in activation induced cell death of physiologically activated T cells. Loss of Fas has been linked to escape from the antitumoral immune response in diverse cancer entities.39 Fas receptor and FasL are transmembrane proteins of the tumor necrosis factor family and are expressed on cells of lymphoid and myeloid lineage. On binding of the Fas ligand to Fas, a cascade of events is initiated that finally leads to apoptosis.
Aberrations of the Fas/FasL pathway have been studied in CTCLs. Immunohistochemical studies of Fas expression in skin biopsy specimens from MF lesions demonstrated loss of Fas expression the in tumor stage, while expression was clearly detectable in early patch and plaques stage.40 In addition to the loss of Fas expression on the cell surface, upregulation of intracellular proteins like cFLIP, which inhibit Fas-initiated apoptosis has been shown in CTCL cells.41 Thus, downregulation or loss of Fas expression as well as disturbance of intracellular pathways activated by death receptors may counteract proapoptotic signals given to the tumor cells by antitumoral immune cells, finally resulting in immune escape of the tumor cells in CTCLs.
In conclusion, specific mechanisms for all features that have been proposed to be necessary for the evolution of tumor cells,42 like evasion of apoptosis, generation of growth or survival signals and insensitivity to antigrowth signals, mechanism of tissue homing and metastasis have nowadays been demonstrated in CTCL cells.
Finally CTCLs exhibit a modulated antitumor response. It is well established that patients with CTCLs have variable production and response to T helper 1 and 2 (Th1, Th2) cytokines. Early studies that measured the messenger RNA (mRNA) levels of Th1 and Th2 cytokines showed that, at any stage of disease, mRNA of the Th2 cytokines interleukin 4 and 5 (IL-4, IL-5), could be found, with the persistence of Th2 cytokine mRNA in skin more frequent in the tumor stage. Correlation of the amount of the Th2 cytokine IL-10, which has immunosuppressive functions, and disease stage in CTCLs has been described.43 The decline in interferon (IFN)-γ and predominance in Th2 cytokines, which is accompanied by a decreasing number of circulating dendritic cells in the blood, might be responsible for the loss of immunosurveillance, which allows CTCL to progress. Lesions of CTCLs often have a CD8+ T-cell component. Although they were once thought to be innocent bystanders in the epidermis, data have now defined the responsibility of CD8+ T cells in the antitumor response. The first insight into defects in the integrity of normal T-cell function in patients with advanced stages of CTCLs came from the observation of patients who had been treated with extracorporeal photochemotherapy (ECP).44 Responding patients had significantly lower CD4/CD8 T-cell ratios and higher absolute numbers of CD8+ T cells in their peripheral blood at the outset of treatment than did nonresponders. In general, skin biopsy specimens of MF lesions in early stages (T1 and T2) harbor a higher proportion of CD8+ cells than lesions in advanced stages. Improved long-term prognosis was correlated with the presence of these CD8+ tumor-infiltrating lymphocytes (TIL) in MF.45 It is now believed that these CD8+ cytotoxic T cells play an important part in the pathogenesis of CTCLs, mainly in mediating an antitumor response.
An interesting hypothesis pursued during the recent years, proposes that the malignant T cells themselves exhibit properties of regulatory T cells (T regs) and thereby are able to modulate the antitumoral immune response.46 However, the results of studies enumerating T regs in CTCL biopsy specimens were inconsistent. These conflicting results may be due to the fact that both a decrease of T regs as well as an increase of CD25-negative T regs, which then are phenotypically identical with the malignant clone, can be observed in SS.47
MF is the most frequent disease among CTCLs, usually arising in mid-to-late adulthood (median age at diagnosis, 55–60 years) with a male predominance of 2:1.
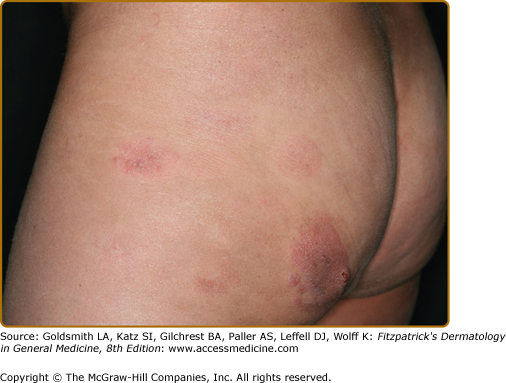
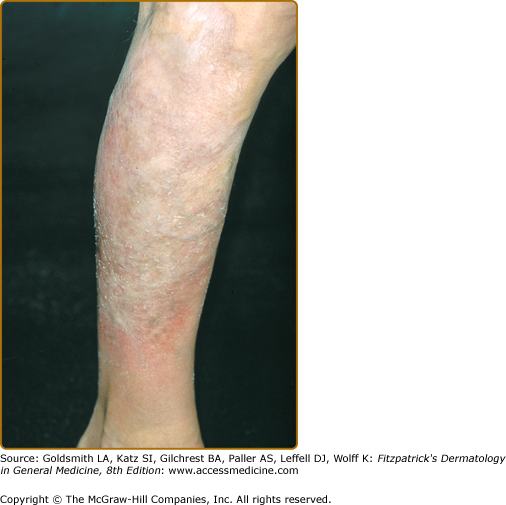
Clinically, MF is categorized as being in the patch, plaque, or tumor stage, but patients may simultaneously have more than one type of lesion. In early patch-stage MF (Figs. 145-1 and 145-2), there are single or multiple erythematous, scaly macules and patches that vary in size and are usually well defined. The color of the lesions may vary from orange to a dusky violet–red. The distribution classically favors nonsun-exposed sites, with the “bathing trunk” and intertriginous areas predominant early in the course of the disease. The eruption may be intensely pruritic or asymptomatic and occasionally may be transitory, disappearing spontaneously without scarring. Diagnosis at this stage may be difficult. Often a patient will recall a preceding “chronic dermatitis” for 10–20 years that may have been considered to be therapeutically resistant contact dermatitis, atopic dermatitis, psoriasis, or eczema. In any patient with a dermatosis that is refractory to the usual modalities of treatment, multiple biopsy specimens should be taken to pursue a diagnosis.
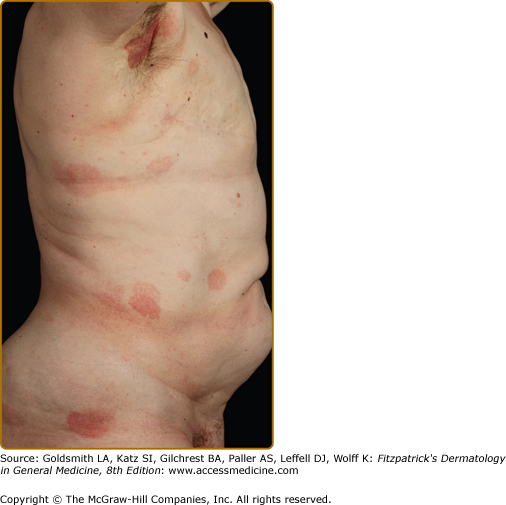
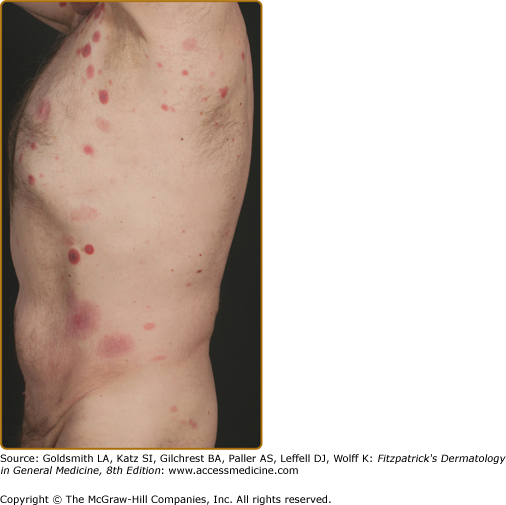
Patches may last for months or years before progressing to the plaque stage (Fig. 145-3), or plaques may arise de novo. Plaques appear as sharply demarcated, scaly, elevated lesions that are dusky red to violaceous and variably indurated (Figs. 145-3 and 145-4). Lesions in this stage may regress spontaneously or may coalesce to form large plaques with annular, arcuate, or serpiginous borders, and may clear centrally with disease activity remaining at the periphery of the lesion. There may be purpuric hyperpigmentation or hypopigmentation and poikiloderma.
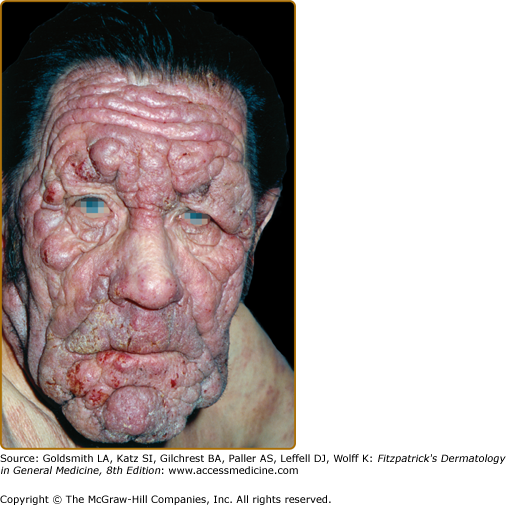
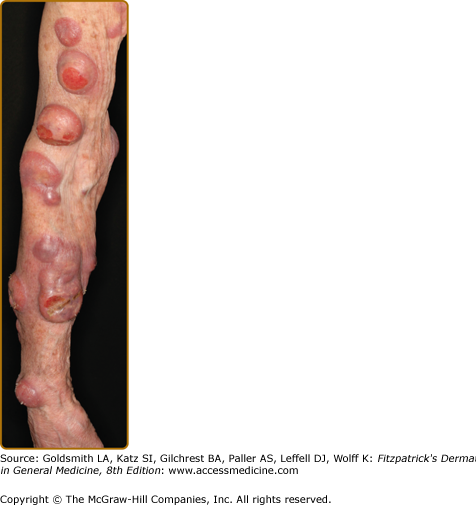
Tumors may occur anywhere on the body, but have a predilection for the face (Figs. 145-5 and 145-6) and body folds: axillae, groin, antecubital fossae, and, in women, the inframammary area. These may occur in preexisting plaques or patches of MF, which coincides with an extension of these lesions in the vertical dimension (Fig. 145-3). At this point, the neoplastic cells behave in a biologically more aggressive manner, with pronounced tumor cell accumulation that leads to the clinical appearance of an expanding dermal nodule (see Fig. 145-6). De novo occurrence suggests metastatic spread by cells of a malignant T-cell clone. The nodules are reddish brown or purplish red and smooth surfaced, but they often ulcerate and may become secondarily infected. Growth rate is variable. Patients with tumors tend to have a more aggressive form of the disease than patients with patch and plaque disease.
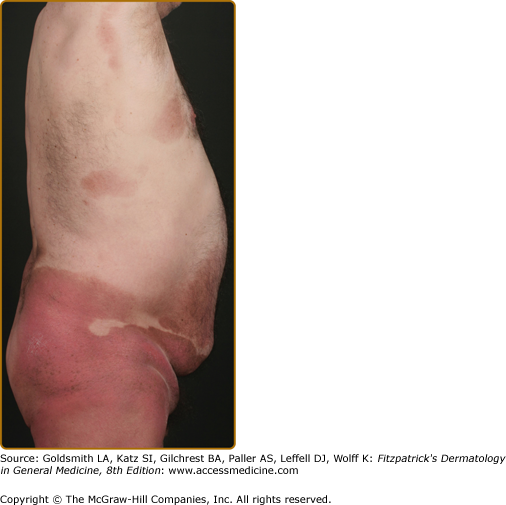
Erythroderma (Fig. 145-7A) may start de novo or develop in MF. The nomenclature for erythrodermic phases of CTCL varies. It has been proposed that erythroderma be defined as the involvement of 80% of body surface area with lesions of ill-defined borders and that patients with a history of preexisting MF be defined as having a separate syndrome of “erythrodermic MF.”
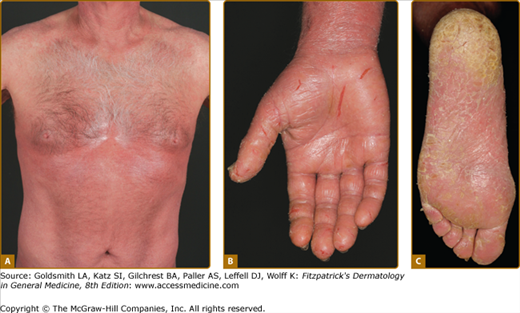
The skin is diffusely bright red with readily apparent scaling, but there may be characteristic islands of uninvolved skin, termed nappes claires (Fig. 145-2). There may be sparing of the areas of skin that are frequently folded, such as the abdomen and antecubital and axillary areas. This sparing produces a finding often called the deck chair or folded luggage sign. Some patients with the erythrodermic form of CTCL develop tumors.
Patients may complain of fever, chills, weight loss, malaise, insomnia secondary to the overwhelming pruritus, and poor body temperature homeostasis. There may be hyperkeratosis, scaling and fissuring of the palms and soles, alopecia, ectropion, nail dystrophy, and ankle edema, with the integument being shiny and hidebound. These changes result in pain on walking and extreme difficulty with tasks requiring manual dexterity. Such patients experience severe restrictions by the extent and localization of their skin manifestations. Pruritus is often intense, which results in excoriation, exudation, and secondary infection that may dominate the clinical picture.
The differential diagnosis of MF is summarized in Box 145-2.
PATCH/PLAQUE STAGE
TUMOR STAGE
ERYTHRODERMA
|
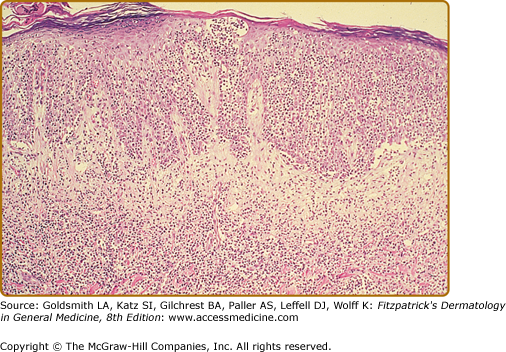
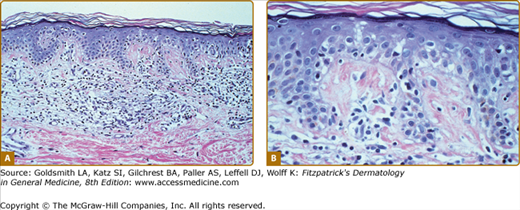
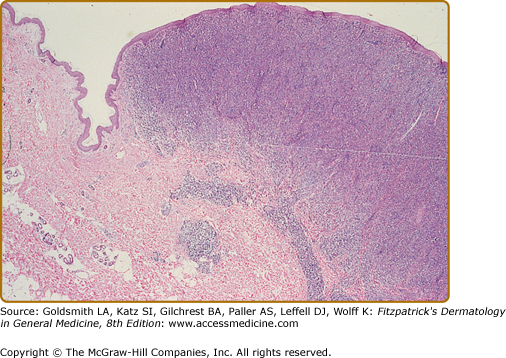
In the patch, plaque, and also in the erythrodermic stage, there is a band-like infiltrate in the upper dermis composed of reactive T cells and neoplastic T lymphocytes, which are characterized by hyperconvoluted cerebriform nuclei. The neoplastic T cells show an epidermotropism with formation of intraepidermal Pautrier’s microabscesses (Figs. 145-8 and 145-9). In the tumor stage, a nodular infiltrate in the dermis is found, and the epidermal component is much less pronounced (Fig. 145-10). Immunohistologically, the malignant cells express a mature peripheral T-cell (CD4+) phenotype. Partial loss of pan-T-cell antigens such as CD7 and CD3 may be a feature of MF but is not pathognomonic of the disease. Analysis of T-cell receptor genes (TCR) typically shows a clonal rearrangement as demonstrated by PCR or Southern blot techniques.
Treatment should be stage-adapted and alleviate symptoms (see Sections “Staging of Cutaneous T-Cell Lymphomas” and “Principles of Treatment of Cutaneous T-Cell Lymphoma”). The prognosis depends on the type and extent of skin involvement (plaques, tumors, or erythroderma), the presence of lymph node involvement, and the presence of visceral disease. Overall, patients with MF limited to the skin have a 5-year survival rate of 80%–100%. In contrast, patients with lymph node involvement show a 5-year survival rate of 40%.
Folliculotropic MF represents a distinct variant of MF characterized by preferential involvement of the head and neck by folliculotropic T-cell infiltrates, with or without mucinous degeneration of the hair follicles (Fig. 145-11B). Previously, this variant was called follicular mucinosis or alopecia mucinosa. Folliculotropic MF affects mostly adults and is rarely observed in children and adolescents. Patients may have grouped follicular papules (see Fig. 145-11A), acneiform lesions, indurated plaques, and sometimes tumors, which usually involve the head and neck region. The occurrence of hair loss within the lesions, most conspicuous on the eyebrows, an intense pruritus, and secondary bacterial infections is common.
Patients with dark skin develop hypopigmented MF, a variant of patch MF. In darker skinned individuals, this may be the most common presentation of the disease. Patients respond to therapy by repigmentation, and the reappearance of hypopigmented lesions often indicates a relapse.
Pagetoid reticulosis (Woringer–Kolopp disease) is a distinct variant of MF that is characterized by the presence of localized patches or plaques with an intraepidermal proliferation of neoplastic cells. Patients present with a solitary psoriasiform or hyperkeratotic patch or plaque, which is usually localized on the extremities (Fig. 145-12) and is slowly progressive. Unlike in classic MF, extracutaneous dissemination has not been observed.
In the past, the term pagetoid reticulosis also included a disseminated form (called Ketron–Goodman type). As this type usually shows a more aggressive behavior, it is now classified as an aggressive primary cutaneous epidermotropic CD8+ T-cell lymphoma (see Section “Primary Cutaneous Aggressive Epidermotropic CD8+ T-Cell Lymphoma”).
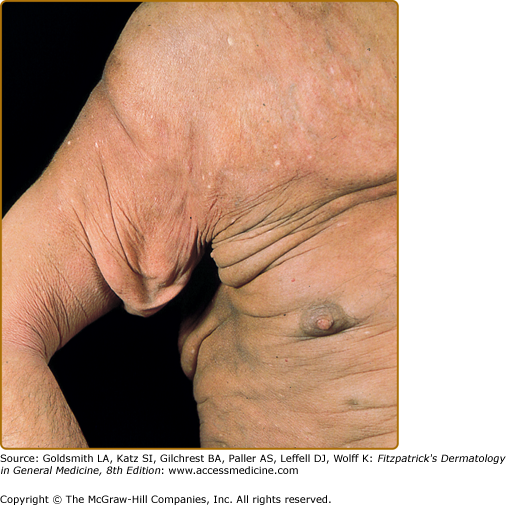
Granulomatous slack skin is a rare subtype of MF characterized by localized areas of bulky folding of the skin, with a predilection for the axillae and groins (Fig. 145-13). Light microscopy reveals a dense granulomatous infiltrate in the entire dermis. In addition to small atypical cells with cerebriform nuclei, macrophages and multinucleated giant cells occur and loss of elastic fibers is observed. The neoplastic cells express a CD3+ CD4+ CD8– phenotype.48
SS is characterized by the triad of diffuse erythroderma, generalized lymphadenopathy, and circulating malignant T cells with cerebriform nuclei, the so-called Sézary cells.
The erythroderma is often accompanied by severe scaling or fissuring of the palms and soles (see Fig. 145-7A–C), alopecia, and onychodystrophy and may be associated with marked exfoliation, edema, and lichenification and an intense pruritus.
SS demonstrates histologic features similar to those of MF, but repeated biopsies may be necessary as specimens often show nondiagnostic findings. Therefore, several diagnostic criteria for the blood involvement proposed by the International Society for Cutaneous Lymphoma—including an absolute Sézary cell count in the peripheral blood of at least 1,000 cells/mm3 detected by light microscopic examination of a blood smear, or an increased CD4/CD8 ratio of more than 10 measured by fluorescence-activated cell-sorting analysis, or evidence of a circulating T-cell clone detected by cytogenetic methods—were included in the WHO–EORTC classification.1 Patients lacking the atypia on the peripheral blood smear or the corroborating evidence are considered to have erythrodermic CTCL.49
Compared with patients with patch- or plaque-stage MF, patients with SS have a markedly decreased 5-year survival rate of 24%. By the time the SS appears, there is very little normal immunity left. Indeed, SS patients often die because of infectious complications.50
Primary cutaneous CD30+ lymphoproliferative disorders consist of LyP and cALCL. Both entities are now recognized to form the ends of a spectrum, which includes borderline cases that, on histomorphologic grounds alone, cannot be clearly assigned to LyP or cALCL.
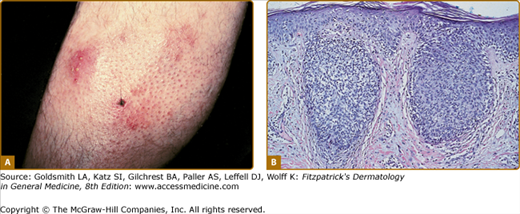
LyP was first described by Macaulay in 1968. It is an uncommon chronic disorder (incidence of 1.2–1.9 cases in 1,000,000) characterized by recurrent self-healing crops of papules and papulonodules.
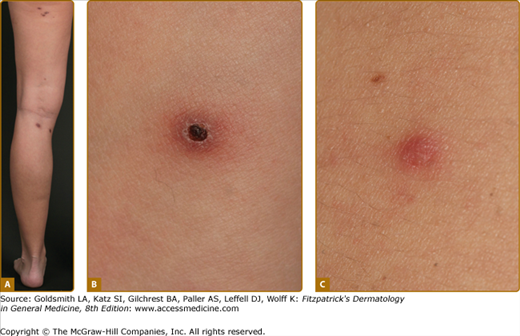
Disseminated recurrent self-healing papules and papulonodules occur that may become necrotic in the center (Fig. 145-14) and often erupt over a period of months or several years. The lesions typically involve the trunk and extremities, and lesions in various stages of evolution may be present concurrently.
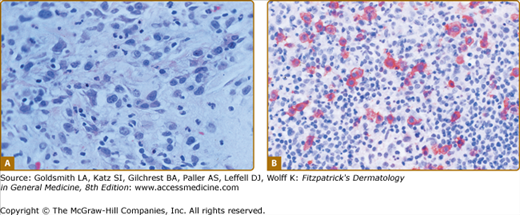
LyP shows a highly variable histologic picture that closely mimics that of malignant lymphoma. Three major histologic types, designated as A, B, and C, have been recognized.51 Types A and C are characterized by the presence of large atypical blasts, including mononucleated and binucleated or multinucleated cells resembling Reed–Sternberg cells characteristic of Hodgkin lymphoma. The atypical blasts express one or more T-cell antigens as well as the lymphoid activation antigen CD30 (Fig. 145-15). Although in type A these cells are embedded in a dense inflammatory background consisting of histiocytes, small lymphocytes, neutrophils, and/or eosinophils, in type C they form large sheets closely simulating cALCL. LyP type B is composed of small-to-medium-sized CD30-negative T cells showing epidermotropism and thus closely resembles classic MF. Importantly, different histologic types of LyP can be present in one patient, because the histologic picture also correlates with the age of the individual lesion.
Because a curative therapy is not available and none of the available treatment modalities affects the natural course of the disease, the short-term benefits of active treatment should be balanced carefully against the potential side effects. Low-dose methotrexate (5–20 mg/week) is the most effective therapy to suppress the development of new skin lesions. Treatment with psoralen plus ultraviolet A light (PUVA) has been reported to yield beneficial effects but duration of response is often short-lived after discontinuation of treatment. Therefore, in patients with few, nonscarring lesions, long-term follow-up without active treatment should be considered.52
In general, LyP shows a benign clinical course and a favorable 10-year survival rate of nearly 100%. However, in a proportion of patients, estimated at 10%–20% of cases, LyP can precede, coexist with, or follow malignant lymphoma, especially MF, Hodgkin lymphoma, or nodal anaplastic large cell lymphoma. In many of these cases, the same clonal TCR rearrangements have been found in the LyP as well as in the associated lymphoma. In the majority of LyP cases, despite the sometimes extremely long course of the disease, there is no evolution of a secondary lymphoma.
The cutaneous lymphoma cALCL is characterized by large tumor cells, of which the majority express the CD30 antigen, with no evidence or history of MF or another type of primary CTCL. Regardless of the morphology of the tumor cells (anaplastic, immunoblastic, or pleomorphic large cells), the clinical presentation and behavior are identical.
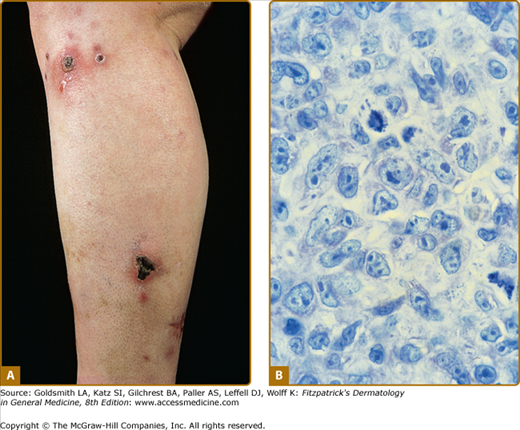
CD30+ cutaneous large-cell lymphomas occur in adults and rarely in children and adolescents, with a male–female ratio of 1.5:1. The clinical picture is characterized by the solitary or locoregional occurrence of reddish to brownish nodules and tumors, which frequently ulcerate (Fig. 145-16A). Although secondary involvement of the regional lymph nodes is observed in roughly 10% of the patients, it is not necessarily associated with an unfavorable prognosis.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


