Cutaneous Langerhans Cell Histiocytosis: Introduction
|
Introduction
The term “histiocytoses” identifies a group of diseases that have in common the proliferation of cells of the mononuclear phagocyte system, including dendritic cells.
Histiocytes, formally connective tissue macrophages, and dendritic cells constitute two of the major types of nonlymphoid mononuclear cells and are involved in immune and nonimmune inflammatory responses. Both cells arise in the bone marrow from pluripotential stem cells, but follow different paths of differentiation (Fig. 147-1). The macrophage, detectable by CD68, is a monocyte in the peripheral blood and a macrophage in tissue. It may differentiate into a number of different giant cells, including foreign body, epithelioid, Touton, and Langhans types.
Figure 147-1
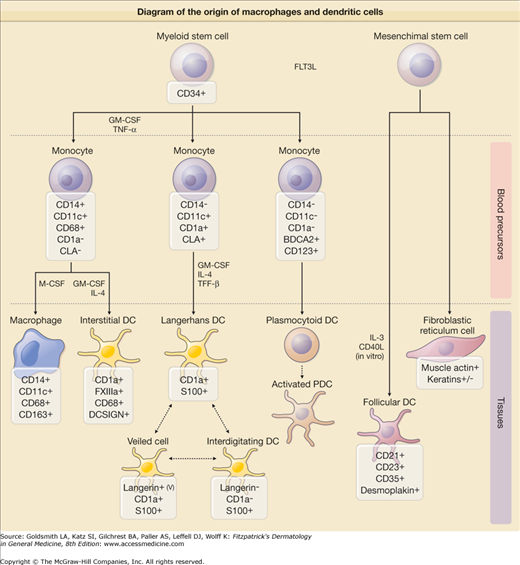
Schematic diagram of the origin of macrophages and dendritic cells. Both macrophages and dendritic cells (antigen presenting cells) are derived from a common bone marrow precursor. In contrast, follicular dendritic cells are thought to be of nonhematopoietic origin. +, most, if not all, cells positive; −, all cells negative, +/−, a minority of cells positive; v, variable intensity. (Adapted from Jaffe R et al: Histiocytic and dendritic cell neoplasms, introduction. In: WHO Classification of Tumors of Haematopoietic and Lympoid Tissues, edited by SH Swerdlow et al. Geneva, Lyon, WHO Press, 2008.)
Dendritic cells are antigen-presenting cells that interact with T cells. The prototype of dendritic cell in the skin is the Langerhans cell (LC). First described by Paul Langerhans in 1868 as a neuronal cell, the LC was first linked to antigen presentation during the 1940s. Precursor cells to the LCs migrate from the dermis into the epidermis where they complete differentiation. LCs have long cytoplasmic projections and a large almost kidney-shaped nucleus. Electron microscopy reveals the characteristic cytoplasmic organelle, the Langerhans or Birbeck granule, a tennis racket-shaped structure that is involved in pinocytosis and receptor-mediated endocytosis. Langerin (CD207), a receptor that permits internalization of antigen into Birbeck granules and presentation of antigen at the cell surface, is the most specific marker for LCs. LCs are also S100- and CD1a-positive. Once LCs have contacted antigen, they move via the lymphatics to the T-cell zones of the regional lymph nodes where they are designated interdigitating reticulum cells. There LCs present antigen to naive T-cells, activating them to proliferate if the antigen is recognized. Indeterminate cells are most likely immature Langerhans cells that lack Birbeck granules. Dermal dendritic cells are perivascular cells that are also capable of antigen uptake and presentation. Dermal macrophages can be identified in the normal dermis but their number increases during inflammation. Dermal macrophages, dermal dendritic cells, and Langerhans cells have a common bone marrow precursor, explaining why there are so many overlapping functions and markers.1
Langerhans Cell Histiocytosis
Langerhans cell histiocytosis (LCH) is characterized by aberrant proliferation of the LCs. Lichtenstein grouped Letterer–Siwe disease, Hand–Schüller–Christian disease, and eosinophilic granuloma of bone as “histiocytosis X” in 1953, based on finding abnormal histiocytes in these three related disorders. During the ensuing decade, Basset and Nezelof investigated novel intracellular granules in the lesional histiocytes and recognized them to be identical to those described in normal LCs. The term Langerhans cell histiocytosis (LCH) is now preferred to “histiocytosis X”.2
Epidemiology
The prevalence of LCH is internationally estimated to be from 4.0 to 5.4 per million population, but these numbers may be low because of failure to recognize the organ and skin involvement in many patients. In children, the annual incidence is estimated at 0.5 per 100,000 children in the United States and at 4 per million worldwide.3 The male–female ratio is 2:1. The disease can occur in individuals of any age and can also be congenital, but is most common in children aged 1–3 years.
Etiology and Pathogenesis
Histiocytoses are caused by a dysregulated proliferation or activation of dendritic cells or macrophages, both belonging to the mononuclear phagocyte system. Despite the improved knowledge of the classification of histiocytic disorders and the establishment of guidelines for diagnosis, treatment, and follow-up provided by the Histiocyte Society, the etiology and pathogenesis of most of these diseases are poorly understood, partially because of the lack of reliable animal models.4
The pathogenesis of LCH remains a mystery despite numerous investigative efforts. In addition to genetic factors, infectious agents (especially viruses), and cellular and immune system dysfunction, including abnormalities of lymphocytes and cytokines (interleukin 1α, interleukin 10), cellular adhesion molecules, and a combination of these, have been implicated. The role of genetics is not well defined. Although LCH has been reported in both monozygotic and dizygotic twins, the relative rarity of the familial occurrence does not favor a significant hereditary influence.
The reactive versus neoplastic nature of LCH cells is also debated. Although clonality has been demonstrated in all forms of LCH, this does not mean that the process is histologically and biologically malignant. The following features substantiate the reactive hypothesis: (1) LCH cells are highly differentiated, (2) histologically the granulomatous lesions resemble those seen in infections or foreign-body reactions, and (3) lesions may spontaneously regress. These pathogenic steps have been suggested: (1) viruses induce activation of histiocytes, (2) immunologic mechanisms produce the pathologic Langerhans cell phenotype, and (3) a cytokine-mediated storm results in LCH lesions. Many viruses (human herpesvirus 6, cytomegalovirus, adenovirus, and parvovirus) are suspected to play an etiologic role, but none has been proven to be responsible for the disease.
In contrast, the following observations support a clonal neoplastic origin of LCH: (1) bone marrow derivation and clonality of LCH cells, (2) the existence of familial cases of LCH, (3) findings of chromosomal instability with an abnormality on chromosome 7 and of p53 protein in LCH, and (4) cooccurrence of LCH and a myelodysplastic marrow. Two possible explanations have been proposed for the association between LCH and myelodysplasia. The first is that the marrow dysplastic features are secondary to the infiltration of the marrow by LCH cells induced by cytokines and do not represent elements of a true myelodysplastic syndrome. A second possibility is that the abnormal marrow cells are related to the clonal LCH cells and are part of the disease process, for example, myelodysplastic cells could be arising from the same clonal cell, an anomalous pluripotent dendritic (stem) cell line. This possibility raises the issue of whether LCH could be considered a “myeloid dendritic stem cell disorder.” These findings also suggest a genetic predisposition in LCH and stimulate the search for potential candidate genes in LCH. Figure 147-1 illustrates the histiocyte developmental pathway according to Jaffe et al.5
Clinical Findings
LCH is a disorder with a broad spectrum of forms: it can be local, asymptomatic with an indolent course, limited to an isolated bone lesion, or systemic and aggressive involving multiple organs and producing significant symptoms. Thus, the clinical manifestations depend on the site of the lesions and the systems involved. Historically, LCH has been described under various names: Letterer–Siwe disease is the prototype of the acute, disseminated, multisystemic form that usually appears in infants or newborns. Its course, if the disease is untreated, is fatal; Hand–Schüller–Christian disease is the chronic, progressive, multifocal form, commonly beginning in childhood; eosinophilic granuloma is the localized, benign form; and Hashimoto–Pritzker disease represents the benign, self-healing variant of LCH, usually present at birth or during the first few days of life. In 1997, experts of the Reclassification Working Group of the Histiocyte Society advocated the disuse of the eponyms. They recommended the use of the term Langerhans cell histiocytosis only and the stratification of patients based on the extent of disease. The classification system for histiocytosis defined by the Histiocyte Society is shown in Table 147-1. At present, it seems realistic to describe LCH as a continuum as in Fig. 147-2, because every patient is unique with regard to the type, number, and location of lesions.
| Single-system Disease | |
| Localized (single site) | Monostotic bone involvementa |
| Isolated skin involvement | |
| Solitary lymph node involvement | |
| Multiple site | Polyostotic bone involvementa |
| Multifocal bone disease (two or more different bones) | |
| Multiple lymph node involvement | |
| Multisystem Disease | |
| Low-risk group | Disseminated disease with involvement of low-risk organs (skin, bone, lymph node, pituitary) |
| High-risk group | Disseminated disease with involvement of one or more of the high-risk organs (hematopoietic system, lungs, liver, and spleen) |
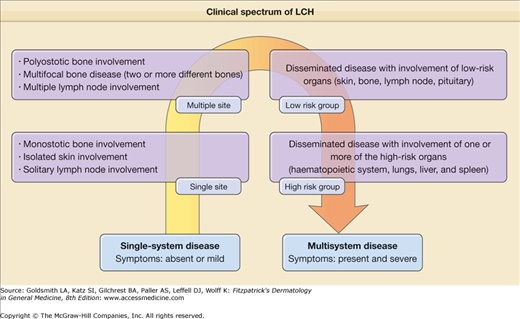
Although the historical terminology can be maintained for didactic purposes, it is more important to distinguish the forms with systemic involvement that require systemic management (multisystemic LCH) from those with localized lesions and the best prognosis that can be treated with a topical medication or observational approach (single-system LCH) or that are “self-healing” (Fig. 147-3).
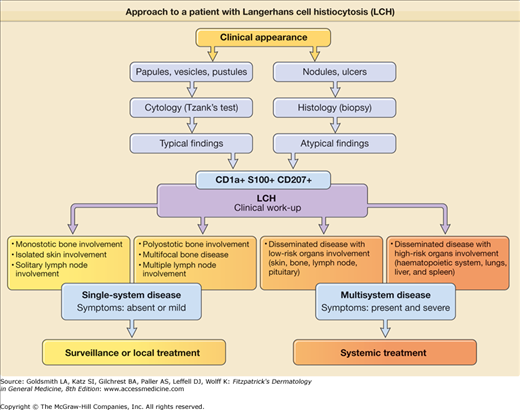
The Writing Group of the Histiocyte Society proposed a guideline identifying confidence levels for the diagnosis of class I histiocytosis (LCH). These criteria were as follows: (1) presumptive diagnosis—light morphologic characteristics; (2) designated diagnosis—light morphologic features plus two or more supplemental positive results to stains for adenosine triphosphatase, S100 protein, α-d-mannosidase, and peanut lectin; (3) definitive diagnosis—light morphologic characteristics plus Birbeck granules in the lesional cell visible with electron microscopy and/or positive results on staining for CD1a antigen on the lesional cell.
Current immunostaining techniques allow the definitive diagnosis of LCH from just paraffin-embedded tissue, which avoids the need for sophisticated and expensive methods such as electron microscopy. Because the location and diffusion of the lesions have a significant influence on the course of the disease and its prognosis, various classification schemes assign scores for each organ and system involved. The former classification of LCH into three or four disease categories is no longer used. The present system attempts to evaluate the extent of involvement and its relationship to prognosis. The former classification of LCH into three or four disease categories is no longer used. The present system attempts to evaluate the extent of involvement and its relationship to prognosis.
Cutaneous manifestations are very common in LCH and may represent the earliest sign of the disease. The typical lesion is a small translucent papule, 1–2 mm in diameter, slightly raised, rose-yellow (Figs. 147-4A and 147-4B), and usually located on the trunk (see Figs. 147-4A and 147-4B) and scalp. The lesions frequently show scaling (see Fig. 147-4A) and may become crusted and ulcerated (see Figs. 147-4B and 147-4C). Vesicles and pustules may occur, simulating eczema, miliaria, scabies, and varicella. This is an especially common presentation during the neonatal period (first month of life). The presence of purpura is a poor prognostic sign. Hashimoto–Pritzker disease has traditionally been characterized by the eruption of multiple or solitary elevated, firm, red–brown nodules (Fig. 147-5A) or flesh-red lesions similar to infantile angiomas; late papulonodular lesions may show elevated borders and ulcerate easily. These lesions can grow in size and number in the first few weeks of life, and some may become quite large. They then form brown crusts, which are shed and occasionally leave whitish atrophic scars after spontaneous resolution. Since the original description of the papulonodular lesions, it is clear that small reddish-brown crusted papules that may resemble chickenpox are also a common manifestation of “self-healing Langerhans cell histocytosis,” with spontaneous clearance by 2–3 months of age (see Fig. 147-5B). Presentation with extensive, erosive, superficial lesions suggestive of congenital bullous epidermolysis has also been described.8
Figure 147-4
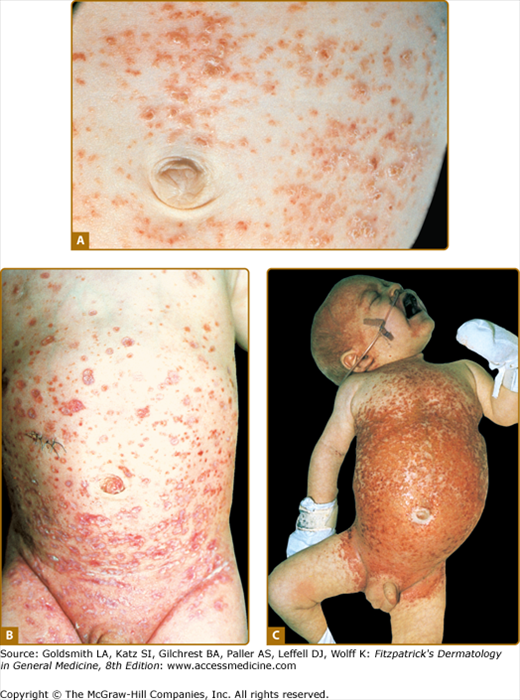
Extensive Langerhans cell histiocytosis. A. Abdominal area of infant with multiple yellowish erythematous papules covered by scale or crust. B. Infant with multiple ulcerated skin lesions on the trunk. C. Severe disease with confluent erosive and crusted lesions forming ulcers; distension of the abdomen reflects hepatosplenomegaly.
Figure 147-5
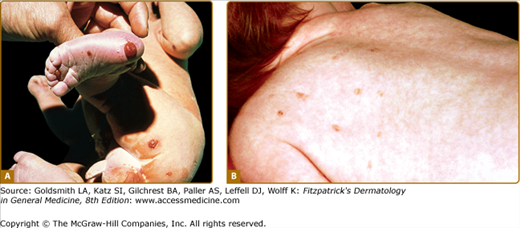
A. Congenital “self-healing” reticulohistiocytosis (also called self-regressive cutaneous LCH and formerly Hashimoto–Pritzker disease). Multiple disseminated, elevated, firm red–brown or dark red nodules (multiple form) present at birth. (From Bonifazi L et al: Congenital self-healing histiocytosis. Arch Dermatol 118:267, 1982, with permission.) B. Resolving crusted reddish brown papules of congenital self-healing Langerhans cell histiocytosis with chickenpox-like lesions.
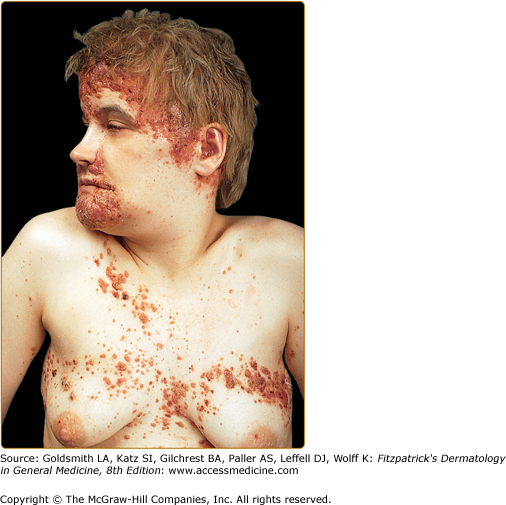
Cutaneous lesions may appear rapidly in successive crops. The lesions tend to merge on the scalp, mimicking seborrheic dermatitis or folliculitis, and leading to alopecia, and on the folds (retroauricular, axillary, and genital), mimicking intertrigo. Occasionally, the lesions merge into plaques on the medial aspect of the chest (Fig. 147-6), the midback, and temporoparietal regions, and may become xanthomatous.
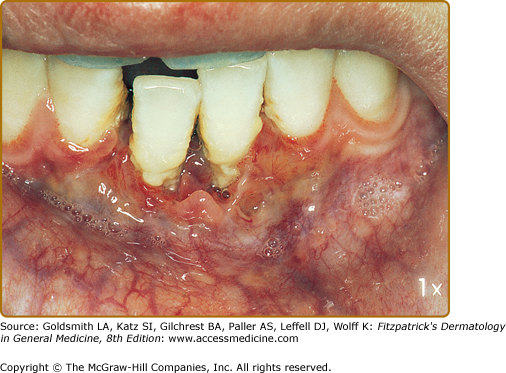
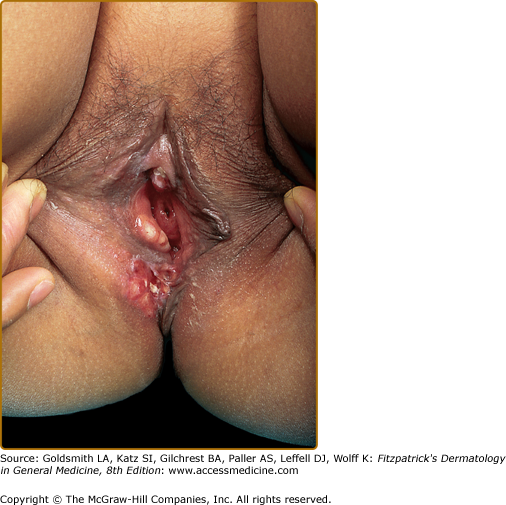
Mucous membrane lesions are commonly noduloulcerative and mainly involve the perioral area and gingiva (Fig. 147-7) and the perigenital or perianal regions (Fig. 147-8). Oral manifestations may be the first sign of LCH: nonspecific pain, aphthae, gingival bleeding, candidiasis, ulceration with loosening of the teeth, and premature eruption of the teeth can be observed.
Nail changes include fragile lamina, paronychia, subungual pustules, nail fold destruction, onycholysis, subungual hyperkeratosis, longitudinal grooving, and pigmented and purpuric striae of the nail bed (Fig. 147-9). These features are considered to be unfavorable prognostic signs. Cutaneous manifestations in elderly patients (Fig. 147-10) are similar to those seen in children.
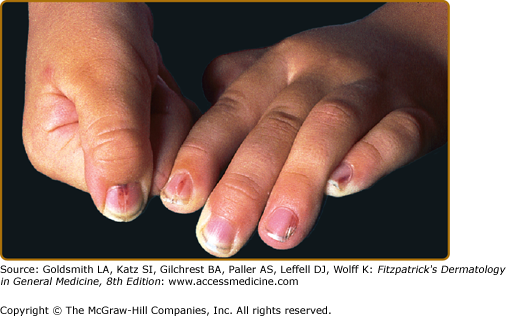
Figure 147-10
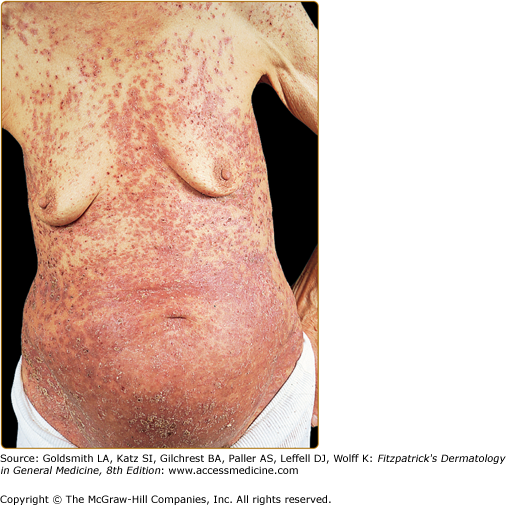
Extensive Langerhans cell histiocytosis in an 81-year-old woman. The diffuse papuloscaling and papulocrusted lesions on the trunk are similar to those seen in early childhood. Systemic involvement consists of hepatosplenomegaly and pulmonary lesions. (From Caputo R et al: Mucocutaneous expressions of Langerhans cell histiocytosis in adults. Eur J Dermatol 13:481, 1994, with permission.)
Malaise, weight loss, failure to thrive, nausea, myalgia, arthralgia, and fever are frequently present in aggressive forms. Bone lesions are the most frequent manifestations of LCH (80% of cases), and bone involvement alone is seen in 50% to 60% of cases. The bones that are most commonly involved by LCH are the skull, femur, mandible, pelvis, and spine. Cervical LCH lesions often extend to paravertebral soft tissue, epidural space, pedicles, and even to the endplate and lamina.9 The lesions of the skull most often involve the temporoparietal region, in which infiltrates lead to limited osteolytic foci that merge to form typical “map” lesions (Fig. 147-11
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


