Cutaneous Changes in Nutritional Disease: Introduction
|
Nutrition is the complex series of events by which living organisms consume and assimilate foods and other nutrients in order to live, grow, and maintain homeostasis. Proper nutrition involves the consumption of key macronutrients in balanced tandem with essential micronutrients. Consisting of carbohydrates, proteins, and lipids, macronutrients are those that are needed in large quantities by an organism to both fuel metabolic processes and provide the substrate for building and maintaining cellular structure. By contrast, micronutrients signify vitamins and minerals, which while necessary to good health, are required in relatively minute quantities. Because humans are unable to synthesize these molecules, clinical disease results when disturbances occur in that equilibrium—most commonly from nutrient deficiencies, but also from an unbalanced ratio of consumed nutrients, or less commonly from nutrient excesses.
With an improved understanding of the role of diet in health and the advent of nutritional supplements, long-standing named scourges such as scurvy, beriberi, and pellagra have largely become diseases of historical interest. Even so, nutritional diseases remain problematic in developing countries and in settings of war, famine, and poverty. In industrialized countries, nutritional diseases may still arise among the disenfranchised: the homeless and those suffering from alcoholism or other forms of substance abuse. Individuals at risk also include those with derangements in their normal diets such as might be encountered with eating disorders or unusual dietary habits as well as with parenteral nutrition. Hypercatabolic states, exemplified by those with cancer, AIDS, hepatic or renal disease, and certain disease states such as carcinoid syndrome, may develop nutrient deficiencies even in the face of normal intake due to increased metabolic requirements. Excessive nutrient losses may occur as a result of decreased absorption arising from gastrointestinal diseases such as cystic fibrosis, inflammatory bowel disease, celiac disease, or following gastrointestinal surgery. Those receiving chronic medications such as anticonvulsants or antibiotics may experience impaired utilization of their nutrients when their medications interfere with gastrointestinal absorption and normal metabolism. Impaired utilization may derive from underlying genetic metabolic defects, enzyme deficiencies, hepatic disease, or drug–nutrient interactions. By contrast, syndromes of nutrient excess generally stem from dietary surplus or iatrogenic therapeutic intake.
Because macronutrients and micronutrients are intrinsically involved in multiple biochemical pathways, disorders of nutrition may produce extracutaneous consequences. Moreover, those at risk for one nutrient deficiency may be suffering from other concomitant deficiencies as well. Appropriate evaluation of the patient with suspected nutritional disease should commence with a detailed history of dietary and medication intake, a review of past medical and family history, and careful inspection of the skin with special attention to the status of hair, nails, and mucous membranes. While some skin findings may be pathognomonic for certain nutritional disorders, physical findings are more oftentimes nondiagnostic. Laboratory analysis of blood and urine nutrient levels may be useful, but poor correlation with tissue levels limits their utility. Radiologic imaging studies may also offer corroboration for diseases such as scurvy, rickets, or beriberi. Clinical improvement following replacement therapy may represent the best or only means to confirm the clinical diagnosis of some nutritional deficiencies. This chapter reviews the important clinical features observed in nutritional disease states.
Macronutrients
Malnutrition is a medical concern of global significance. The United Nations estimates that 1.02 billion individuals worldwide were malnourished in 2009.1 The most recent approximation of child malnutrition estimates that 150 million children under the age of 5, or a quarter of children in this age group, suffered from malnutrition. Approximately 5 million children under the age of 5 die from malnutrition each year.2
In developing countries, malnutrition most commonly arises as a result of inadequate dietary intake, often compounded by disease. War, famine, and poverty states often aggravate the inadequate access to food, which may arise from political unrest, natural disasters, infectious diseases, seasonal and climactic factors, insufficient food production, lack of education, poor sanitation, regional and religious practices, and the limited availability of health care.3 The availability of food in these settings may be limited to diets of corn, rice, and sometimes beans, which provide inadequate amounts of macronutrients, vitamins, and minerals. In Western industrialized countries such as the United States, less than 1% of children nationally suffer from protein–energy malnutrition (PEM). When it is encountered, chronic illness, malabsorption, presumed food allergies, food aversion, nutritional ignorance, and fad diets are more typical etiologies.
PEM refers to a spectrum of disorders describing varying degrees of protein and calorie deficiency. Several subtypes of PEM have been defined on the basis of relative deficiencies in protein and total calorie intake.
Children with marasmus are defined as those with severe wasting and stunting and are at less than 60% expected body weight for age. The term marasmus derives from the Greek marasmos meaning wasting. These changes are the result of chronic and global nutrient deficiencies, often because of a lack of available food. Children with kwashiorkor have body weights less than 60%–80% expected for age, generally as a result of being fed grain-derived foods without adequate accompanying protein or fat. This might occur in geographic areas where grains are more plentiful, but more expensive proteins and fats are not. In fact, the term kwashiorkor derives from the Ghanese term for “the one who is deposed,” referring to the child who is weaned off of breastmilk onto a carbohydrate-rich but often protein-poor diet when the next child is born. The exclusive use of rice beverages, so-called “rice milk” products, as a substitute for baby formulas—either because of their lower cost or because of their perceived hypoallergenicity—has been linked to the development of kwashiorkor among infants in the United States and other Western countries.4–6 A hybrid form of malnutrition in which stunting is associated with edema has been termed marasmic kwashiorkor.
The pathophysiology of these disorders can also be conceptualized as adapted and nonadapted forms of starvation. In adapted starvation (marasmus), decreased intake of all macronutrients, particularly carbohydrates, results in suppressed insulin production. As a result, catabolic hormones act unopposed and allow the appropriate conversion of glycogen into glucose. In the early stages of adapted starvation, muscle breakdown occurs within the first 24 hours, which permits gluconeogenesis to release glucose into the systemic circulation. Later, fat breakdown creates ketone bodies, which can also be utilized by the brain and central nervous system. This reduces the need for further muscle breakdown and therefore ammonia synthesis, so that lean body mass and some protein synthesis can be spared. In prolonged states of adapted starvation, wasting occurs and lean body mass is eventually utilized when all other sources are expended; in the absence of additional nutrient intake, the organism dies. In nonadapted starvation states (kwashiorkor), an imbalance results when intake of carbohydrate is increased relative to decreased intake of protein and fat. In this setting, insulin production is not appropriately suppressed. Without concomitant fat and protein intake, insulin inhibits protein synthesis. Hypoproteinemia, edema, and diarrhea develop, and without protein synthesis, affected individuals are unable to manufacture lipoproteins so fats accumulate resulting in a fatty liver; more importantly, necessary immune proteins are not produced so patients become susceptible to opportunistic infection and septicemia, which represent major causes of mortality in these patients.
While the concepts of adapted and nonadapted starvation provide convenient explanations for why some children develop marasmus and others develop kwashiorkor, some controversy exists. For instance, aflatoxins have been detected with greater frequency in patients with kwashiorkor than in those with marasmus.7 Also, the role of oxidative and nitrosative stress in kwashiorkor is supported by data showing that patients with edematous PEM have lower levels of erythrocyte glutathione and total plasma antioxidants than healthy controls.8
Childhood is marked by periods of significant and rapid growth and development, and nutritional deprivation often manifests as alterations in these patterns of normal growth and development. Failure to thrive, a key finding in patients with PEM, may appear first as wasting in which patients have poor weight gain, and eventually, as decreased rates of linear growth (stunting). Various measurements have been recommended as markers for malnutrition, and include: body weight and length/height relative to age, body mass index, triceps or middle upper arm circumference, as well as skin and hair characteristics.9,10 Suboptimal neurodevelopmental outcomes, heightened susceptibility to infection, and increased mortality may also be observed in chronically malnourished children.
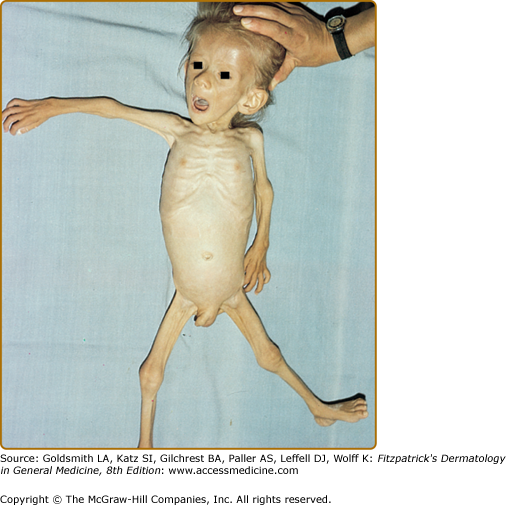
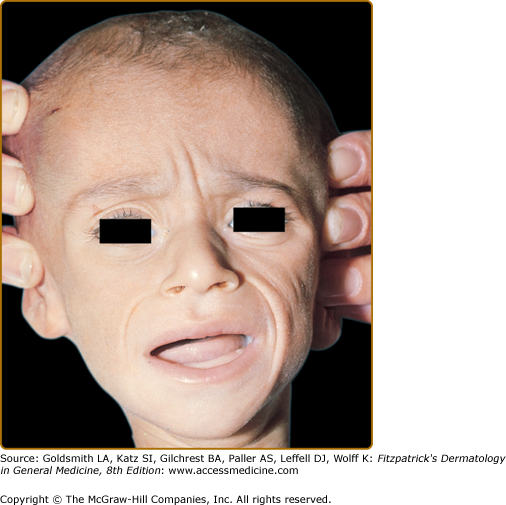
Marasmus typically affects infants under 1 year of age. The physical findings of marasmus include dry, thin, loose, wrinkled skin resulting from loss of subcutaneous fat with an emaciated appearance (Box 130-1 and Fig. 130-1).11 Hair growth slows and examination may reveal easy hair loss leading to thin, fine, brittle hair and alopecia. Increased lanugo hair may also be present. Nails may also show signs of fissuring with impaired growth.12 As the body mobilizes all endogenous energy stores to survive, both subcutaneous fat and muscle mass are lost. The loss of buccal fat pads creates the aged or wizened appearance attributed to children with marasmus that has been referred to disparagingly as “monkey facies”(Fig. 130-2). Perianal fat loss can lead to rectal prolapse and abdominal muscle hypotonia may result in abdominal distention.13 Constipation may alternate with periods of diarrhea that may or may not be associated with concomitant gastrointestinal infection. Angular cheilitis and mucous membrane changes have also been observed in patients with marasmus.14 Patients often show both decreased resting body temperature and bradycardia.
|
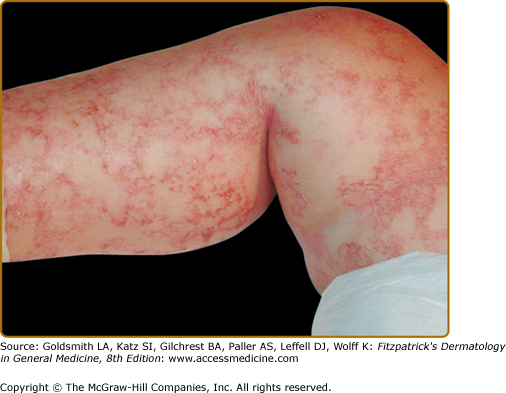
The hallmark features of kwashiorkor—also known as edematous or “wet” PEM—include failure to thrive in association with edema and is primarily noted in children between 6 months and 5 years of age (see Box 130-2). Children are often irritable, but may become lethargic and apathetic. In contrast to marasmus, skin findings are common in kwashiorkor. The generalized dermatitis in edematous PEM has been likened to flaking enamel paint, with the pattern of skin fissuring suggesting cracked or “crazy” pavement (Fig. 130-3). Increased pigmentation of skin may be observed on extensor surfaces of the arms and legs. Hair often changes from its natural color, typically developing a red tint before further pigment dilution results in light, gray–white hair (Fig. 130-4B). If a child experiences intermittent periods of kwashiorkor and improved nutrition, alternate bands of light- and dark color within the hair shaft may be observed and has been referred to as the “flag sign” (Fig. 130-4A). Increased lanugo hair can also be noted in kwashiorkor. In addition to the peripheral edema, a direct consequence of hypoproteinemia, distention of the abdomen is noted as a result of fatty infiltration of the liver.
|

Adults may also be at risk for PEM, particularly those suffering from chronic illnesses, eating disorders such as anorexia nervosa, and the elderly. The prevalence of PEM in adult dialysis patients has been estimated at 25%–50% and is posited to occur as a result of semistarvation with inadequate caloric and protein intake, possibly complicated by the presence of a chronic systemic inflammatory response to dialysis.15,16 Cases of macronutrient PEM have been reported as a complication of bariatric surgery.17 The manifestations of PEM in adults may be less prominent, manifesting more as xerosis or acquired ichthyosis and may be the result of decreased sebaceous gland secretions or concurrent micronutrient deficiency.18 Hyperpigmentation at characteristic sites may also be present, including the perioral, periocular, and malar areas. Diffuse telogen effluvium, lanugo, and thin, dry, dull hair has also been reported in adult PEM.19
Initial laboratory testing in PEM recommended by the World Health Organization20 includes screening for hypoglycemia, anemia, and an investigation into potential comorbid infectious diseases. Since patients with PEM are at significantly increased risk of serious infection, examination of urinalysis for bacterial infection, blood smear for malaria parasites, and feces for blood, ova, and parasites are essential. Skin testing for tuberculosis should be performed. Chest radiography may demonstrate concomitant signs of bacterial pneumonia, tuberculosis, heart failure, rickets, and fractures. Patients with kwashiorkor will also demonstrate hypoproteinemia, which may help guide treatment and prognosis. Evaluation of electrolyte levels before treatment should be interpreted with caution, as refeeding will likely alter mineral balances.
Elevated soluble CD14 levels have been associated with indicators of protein energy wasting, such as low body mass index and muscle atrophy, and increased mortality in hemodialysis patients. CD14 is a coreceptor that triggers immune activation in response to various ligands, including endotoxins and bacterial products. In patients with chronic renal failure on hemodialysis, increased soluble CD14 is associated with elevated markers of systemic inflammation, such as C-reactive protein, interleukin 6, fibrinogen, and plasma pentraxin 3. This suggests a possible link between chronic inflammation, PEM, and mortality in hemodialysis patients.21
Histologic evaluation is generally unnecessary since the clinical features in association with an appropriate history are diagnostic. However, histologic evaluation of the skin reveals increased thickness of the stratum corneum, atrophy of the granular layer, increased basal layer pigmentation, reduction of collagen fibers, and crowding of elastic fibers. Atrophy of the hair appendages with sparing of the sweat gland apparatus can also be noted.22
(See Box 130-3)
Patients with severe PEM often require hospitalization due to the concurrent risks of hypoglycemia, hypothermia, dehydration, and sepsis. Individuals who are not awake and responsive may require intravenous hyperalimentation during the initial stages of therapy; care must be taken to avoid excessively rapid rehydration due to the risks of congestive heart failure. Oral refeeding is generally preferred using oral rehydration salts containing a mixture of essential electrolytes at least until diarrhea subsides or fortified formulas as soon as these can be tolerated. Since severely malnourished children are relatively immunocompromised, empiric antibiotic therapy may be considered on admission for suspected sepsis, and any identified infections should appropriately addressed.20
|
Naturally occurring essential fatty acids (EFA) deficiency states are uncommon in humans. Cases of deficiency arise instead from inadequate intake, malabsorption, or excessive loss. In the past, parenteral nutrition was a common cause of EFA deficiency, but with the introduction of lipid supplementation during parenteral nutrition in 1975, the incidence of EFA deficiency has decreased substantially. Patients at risk for EFA deficiency include those with poor dietary intake, including alcoholics and patients with anorexia nervosa, or those with malabsorptive conditions such as biliary disease, inflammatory bowel disease, postgastrointestinal surgery (e.g., bariatric surgery), and may represent one of the primary etiologies for the rash observed in cystic fibrosis patients.23 Premature low birthweight infants are born with inadequate EFA stores and are also at risk.
EFA represent a group of 18-, 20-, or 22-carbon polyunsaturated fatty acids that cannot be synthesized de novo by the human body. The ω-3 series of fatty acids is found in fish oils, and is derived from α-linoleic acid. The ω-6 series is found in vegetable oils, and is derived from linoleic acid.11 Linoleic acid and α-linoleic acid are the two EFA– that serve as precursors for other EFA and therefore must be obtained from dietary intake. In cell membranes, EFAs increase lipoprotein unsaturation to modulate cell membrane fluidity. Arachidonic acid, a derivative of linoleic acid, is converted into prostaglandins, eicosanoids, and leukotrienes. In the epidermis, linoleic acid contributes to lamellar granule formation in the stratum corneum. Therefore, EFAs play a number of key roles in maintaining homeostasis.
Skin manifestations of EFA deficiency include: xerosis and scaly, diffuse erythema, and associated intertriginous erosions. Poor wound healing, traumatic purpura secondary to capillary fragility, brittle nails, and alopecia may be observed. Affected individuals may also demonstrate hyperpigmentation or hypopigmentation of the hair (Box 130-4). Extracutaneous findings include fatty liver infiltration, increased susceptibility to infection, a blunted immune response, anemia, thrombocytopenia, and growth retardation.
In EFA deficiency states, linoleic acid levels are low and the enzymes that normally convert linoleic acid to arachidonic acid use oleic acid to create an abnormal byproduct. Laboratory evaluation would therefore demonstrate decreased levels of linoleic and arachidonic acids, and elevated plasma levels of an abnormal intermediary, 5,8,11-eicosatrienoic acid. Measurements documenting an increased ratio (≥0.2) of this abnormal intermediary relative to arachidonic acid are diagnostic for EFA deficiency.
While topical application of sunflower seed and safflower oils that contain linoleic acid may improve the clinical cutaneous findings of EFA deficiency,24 topical absorption is unpredictable25 and optimal treatment typically consists of oral or intravenous supplementation of EFA. In order to prevent EFA deficiency, EFA should represent 1%–2% of total daily calories.26
Micronutrients
|
Vitamin A is a fat-soluble vitamin important in retinal photoreceptor function, epithelial proliferation, and keratinization. The two most clinically important metabolites of vitamin A are retinal, which is a key component of rhodopsin generation, and retinoic acid, which regulates cell differentiation.27 Dietary intake of vitamin A derive from both plant and animal sources. Plant sources include dark, green, leafy vegetables, red palm oil, and brightly colored fruits such as papaya, mango, carrots, tomatoes, apricots, and cantaloupe. In plants, the vitamin A precursor β-carotene can be found as a two-molecule complex of the carotenoid known as retinal. The retinal can be later reduced to retinol in the intestinal villous cells. Animal sources of vitamin A include egg yolk, liver, fish, fortified milk, and other dairy products. In animal sources, vitamin A exists as retinyl esters, which are then hydrolyzed to retinol in the intestinal lumen and then absorbed into intestinal mucosal cells. All retinol vitamin A alcohol is esterified to retinyl esters within the intestinal mucosa, released into the bloodstream bound to chylomicrons, and then transported to the liver for storage. Here, retinol can be stored as retinyl esters in the liver; when needed, this storage form may be converted to retinol and bound to retinol binding protein and transthyretin and circulated throughout the body.
Vitamin A deficiency (VAD) can result in cutaneous as well as ocular complications. It is, in fact, the most common cause of preventable blindness in children according to the World Health Organization. VAD has also been associated with defects in immune regulation.
The primary causes of VAD continue to be inadequate intake, fat malabsorption states, and liver disease. In the United States, inadequate intake can be seen in individuals with eating disorders, restrictive diets, and chronic illness. Since vitamin A is fat-soluble, conditions associated with malabsorption of fat such as pancreatic or biliary tract disease, celiac disease, Crohn disease, Shwachman–Diamond syndrome, cystic fibrosis, cholestatic liver disease, chronic intestinal parasitic infection, and gastric bypass surgery can predispose to VAD.
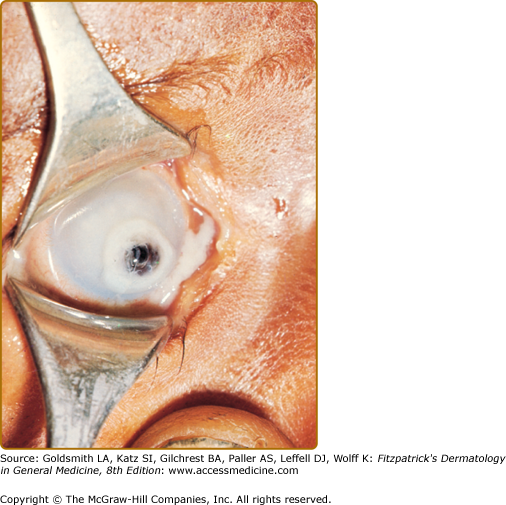
(See Box 130-5). The earliest manifestations of VAD are ocular changes. Impaired dark adaptation (nyctalopia), is followed by xerophthalmia, and as corneal keratin desquamates and overgrowth of Corynebacterium xerosis on the sclera occurs, white patches known as Bitot spots develop. Severe deficiency may lead to corneal xerosis, ulceration, and keratomalacia, which may result in corneal perforation, prolapse of the iris, and blindness (Fig. 130-5).
|
The cutaneous findings of VAD are the result of abnormal keratinization. Mild deficiency may manifest as xerosis and scaling, while more severe deficiency may result in deep skin fissuring referred to as dermomalacia. Squamous metaplasia of the salivary glands as well as the nasal and oral mucosa may occur, leading to xerostomia, hyposmia, and hypogeusia. Laryngeal, bronchial, and vaginal mucosa can also become involved.
Phrynoderma, “toad skin,” (Greek for toad + skin) is typically associated with VAD. These keratotic follicular papules often first develop on the anterolateral thighs and posterolateral upper arms, which then spread to extensory surfaces of the extremities, shoulders, abdomen, back, buttocks, face, and posterior neck (Fig. 130-6). Lucius Nicholas noted the association between this hyperkeratotic folliculitis or phrynoderma with VAD in 1933 when he observed these cutaneous findings among East African workers who developed night blindness and xerophthalmia.28 While originally reported in association with VAD, phrynoderma is a nonspecific finding that can be observed with deficiencies in B-complex vitamins, vitamins C, E, as well as essential fatty acid deficiency, PEM, and general malnutrition states.29
Vitamin A levels can be measured from serum. Normal serum levels are between 20 and 50 μg/dL. Recently, assessment for the hydrolysis of retinoyl glucuronide to retinoic acid has shown promise as an adjunctive test for VAD. Retinoyl glucuronide is a water-soluble form of vitamin A that is not absorbed or hydrolyzed to retinoic acid in vitamin A-replete humans. The presence of serum retinoic acid for 4 hours after oral administration of retinoyl glucuronide was correlated with low serum retinol.30
The recommended daily allowance (RDA) of vitamin A is between 1,000 and 5,000 IU, with younger individuals requiring a lower intake of vitamin A. Recommended treatment for VAD is 100,000–300,000 IU of oral vitamin A daily until symptoms resolve and serum levels normalize.
(See also Chapter 228)
In 1856, Elisha Kent Kane published his two-volume Arctic Explorations, which included accounts of vitamin A toxicity that resulted after his team ingested polar bear liver during his expedition. The toxic substance in polar bear liver was later identified as vitamin A. Since that time, studies have shown that animal livers contain exceptionally high amounts of vitamin A.
Vitamin A toxicity is the result of excess intake of vitamin A and can occur on an acute or chronic basis. Acute toxicity occurs when excessive amounts of vitamin A are ingested over a period of several hours or days. Toxicity typically results when intake exceeds 20 times the RDA in a child or 100 times the RDA in an adult. Chronic toxicity results from daily ingestion of greater than 25,000 IU for more than 6 years or greater than 100,000 IU for more than 6 months of preformed vitamin A. Children appear to be more sensitive to vitamin A intake than adults. Individuals most at risk for toxicity include patients taking systemic vitamin A derivatives for the treatment of dermatologic conditions such as acne, psoriasis, and ichthyosis. The other population at risk includes vitamin food faddists who consume large quantities of nonprescription vitamin A supplements.31 Two notable episodes of vitamin A toxicity occurred in the 1950s when very high levels of vitamin A supplementation were added to infant formulas and the 1970s when high doses of vitamin A were used to treat a variety of dermatologic diseases.32
Recently, vitamin A derivatives have been studied in chemoprevention of keratinocytic carcinomas, such as squamous cell and basal cell carcinomas. A large, blinded randomized controlled study of elderly men with a history of two keratinocytic carcinomas in the 5 years prior to initiation of the study compared topical tretinoin 0.1% cream to placebo. Surprisingly, the study was terminated 6 months early because of a statistically significant increase in all-cause mortality in the tretinoin group compared to the placebo group.33 Analysis of this increased risk was limited by its post hoc nature and suggests that further studies are needed to clarify this association.
(See Box 130-6). Individuals with acute vitamin A toxicity have dry, scaly skin, with large areas of desquamation and fissuring of the lips and angles of the mouth. Other signs and symptoms include headache, fatigue, anorexia, nausea, vomiting, blurred vision, pseudotumor cerebri, myalgias, and arthralgias. An early cutaneous sign of chronic vitamin A toxicity in adults is dryness of the lips, which may progress to diffuse, dry, pruritic, scaly skin with peeling of palms and soles, alopecia, follicular hyperkeratosis, and hyperpigmentation of the face and neck. Anorexia, fatigue, and weight loss may also occur. It is interesting to note that follicular hyperkeratosis may occur in the settings of both VAD and toxicity.
|
In children, chronic toxicity presents as coarse hair with diffuse alopecia, coarse skin with generalized exfoliation, hyperpigmentation, and exfoliative cheilitis. Associated pseudotumor cerebri with headaches and papilledema, and in infants may a bulging fontanelle may be present. Skeletal changes are common with vitamin A toxicity, and may present with growth retardation secondary to premature closure of the epiphyses and spontaneous bone fractures. Proposed mechanisms for the pathologic bone findings seen in vitamin A toxicity involves antagonism between vitamin A- and vitamin D-mediated intracellular signaling pathways and interactions with calcium-regulating hormones.19,31
Laboratory findings in patients with hypervitaminosis A include elevated levels of calcium and alkaline phosphatase. This alteration in calcium homeostasis can lead to calcification of tendons, ligaments, and soft tissues. Deposition of excess vitamin A in adipose tissue and perisinusoidal fibrosis of the liver, which can lead to cirrhosis, are the most significant effect of long-term vitamin A toxicity.
Almost all of the symptoms of vitamin A toxicity subside after the excess vitamin intake is discontinued, with the exception of liver cirrhosis and consequences of pseudotumor cerebri.
While hypervitaminosis A is a disease that causes a broad spectrum of clinical findings, excessive intake of carotene results in a benign disorder characterized by yellow–orange skin pigmentation. The condition was described as “carotenemia” in 1919 by Hess and Meyers who reported a connection between yellow skin pigmentation and increased serum carotene levels.34 During World War I and World War II, carotenemia was more commonly seen because of the dietary shift from a meat-based diet to a more plant-based diet due to food shortages.
As antioxidants, carotenoids have also been studied in cancer prevention. Interestingly, β-carotene supplementation of 20–30 mg per day was associated with an increased risk of lung and gastric cancers.35,36 β-Carotene supplementation is also associated with an increased risk of aggressive prostate cancer.37 Animal studies suggest that excessive carotenoids may increase cyclic AMP signaling and cause deleterious effects on oxidative stress pathways, leading to the increased risk of malignancy.38,39
Carotenes are not synthesized endogenously and are obtained through intake of carotene-rich foods. Plant carotenes are converted to vitamin A in the gastrointestinal tract, but approximately one-third of carotene is directly absorbed. Several factors can affect carotene absorption, including thyroid hormone, pancreatic lipase and bile acid concentrations, processing of foods, and dietary fat and fiber content. Hypothyroid patients notice an elevation of carotene levels as a result of decreased conversion to retinol. Pancreatic lipase and bile acids digest carotene so that a deficiency of these enzymes due to pancreatic or biliary or hepatic dysfunction could result in elevated carotene levels. Mashing, cooking, and pureeing fruits and vegetables increase the availability of carotene because cell membranes are ruptured in the process. Dietary fiber decreases absorption. Because carotene is fat-soluble, a high-fat meal increases absorption. Patients with conditions that lead to hyperlipidemia, like diabetes mellitus, nephrotic syndrome, and hypothyroidism, also predispose patients to carotenemia because of a linear relationship between the amount of β-lipoprotein and carotene. Impaired conversion of carotene to vitamin A in patients with hypothyroidism and liver disease further contributes to carotenemia. Some patients with anorexia nervosa can present with carotenemia because of increased intake of vegetables. Other groups at risk for carotenemia are food faddists, those with excessive intake of nutritional supplements, dried seaweed (nori), carrots, and papayas, and infants ingesting a large amount of pureed vegetables.40–42
Excessive ingestion of carotenes does not result in hypervitaminosis A because the slow conversion of carotene to vitamin A in the intestinal mucosa is not rapid enough to produce toxic amounts of vitamin A.
Carotene deposits in the stratum corneum because of its high lipid content. The yellow discoloration of skin secondary to carotenemia is called carotenoderma. The carotene is excreted by sebaceous glands and in sweat, so the yellow pigmentation appears first on the face, predominantly in the nasolabial folds and forehead, and then progresses to manifest diffusely, especially on the palms and soles. The pigmentation is particularly noticeable in artificial light. Of note, carotenoderma, in contrast to jaundice, spares mucous membranes, like the sclera.
Carotenemia does not occur until serum levels reach three to four times normal levels, greater than 250 μg/dL, and is detectable approximately 4–7 weeks following initiation of a carotenoid-rich diet.
Treatment involves discontinuation of excessive carotene intake, and carotenoderma typically fades as the intake of carotene decreases.
Vitamin D is essential for regulation of calcium and phosphorus metabolism. Vitamin D acts on the gastrointestinal tract to increase dietary calcium and phosphate absorption, stimulates increased bone resorption of calcium and phosphate, and stimulates the renal tubules to increase reabsorption of calcium and phosphate.
Humans obtain vitamin D from two sources: (1) dietary intake, and (2) synthesis in the skin from exposure to ultraviolet light. Common dietary sources of vitamin D include fortified milk, fish oil, and fishes such as salmon, sardines, herring, tuna, cod, and shrimp. Vitamin D can also be synthesized in the epidermis from the precursor molecule 7-dehydrocholesterol (provitamin D3) by ultraviolet light in the 290–320 nm range. Previtamin D3 then undergoes a spontaneous, temperature-dependent isomerization to vitamin D3 (cholecalciferol), which enters the dermal capillaries. At this point, endogenous vitamin D3 joins with exogenous D2 (ergocalciferol) for hydroxylation in the liver to 25-hydroxyvitamin D. This molecule travels to the kidney where it is again hydroxylated to make mature vitamin D (1,25-hydroxyvitamin D, also known as calcitriol).
The most common disorder seen with vitamin D is vitamin D-deficient rickets related to decreased dietary intake of vitamin D. Several genetic causes of rickets also deserve mention. Two types of vitamin D-dependent rickets have been described. Type I represents an autosomal recessive defect in renal vitamin D-1α-hydroxylase, and is therefore treated with supplements of 1,25-hydroxyvitamin D. Type II, also referred to as hereditary vitamin-D resistant rickets, is associated with a rare autosomal recessive end-organ resistance to physiologic levels of 1,25-hydroxyvitamin D. Supplementation with high doses of 1,25-hydroxyvitamin D and calcium may overcome this resistance.
A surge in interest regarding the multisystem effect of vitamin D has spurred numerous studies. Evidence suggests that vitamin D deficiency is associated with increased systolic blood pressures,43 fasting plasma glucose and insulin concentrations,44 risk of cardiovascular disease,45–47 risk of hip fractures in postmenopausal women,48 and colon cancer mortality.49 Vitamin D deficient individuals have an increased rate of all cause mortality when compared to those who are vitamin D replete.47,50 Studies into the function of vitamin D in the immune system have indicated that vitamin D is involved in the innate immune response. Toll-like receptors (TLR) activation triggers expression of vitamin D receptor and vitamin D-1-hydroxylase, which promotes macrophage activation.51 A low vitamin D level is associated with an increased risk of active Mycobacterium tuberculosis infection.52
 Bony deformities in children have been described by artifacts dating back to the first and second centuries c.e. The earliest classic descriptions of the clinical symptoms of rickets appeared separately in 1645 by Daniel Whistler and in 1650 by Francis Glisson. These two men described children in southwest England, where rickets was endemic. At the time they did not understand its etiology. As early as 1824, cod liver oil was noted to help cure rickets. In 1861, Trousseau of France hypothesized that rickets was caused by inadequate diet and sun exposure, but Theodor Palm was the first to study and conclude that there was also a connection between rickets and lack of sun exposure. During the Industrial Revolution, a combination of poor diet, smog and tall buildings obscuring the sun, as well as inadequate time outdoors all contributed to the persistence of rickets in the United States and Europe. By 1918, John Howland, Edward Park, and Paul Shipley had used rat models of rickets to identify the anti-rachitic molecule in cod liver oil, vitamin D.53
Bony deformities in children have been described by artifacts dating back to the first and second centuries c.e. The earliest classic descriptions of the clinical symptoms of rickets appeared separately in 1645 by Daniel Whistler and in 1650 by Francis Glisson. These two men described children in southwest England, where rickets was endemic. At the time they did not understand its etiology. As early as 1824, cod liver oil was noted to help cure rickets. In 1861, Trousseau of France hypothesized that rickets was caused by inadequate diet and sun exposure, but Theodor Palm was the first to study and conclude that there was also a connection between rickets and lack of sun exposure. During the Industrial Revolution, a combination of poor diet, smog and tall buildings obscuring the sun, as well as inadequate time outdoors all contributed to the persistence of rickets in the United States and Europe. By 1918, John Howland, Edward Park, and Paul Shipley had used rat models of rickets to identify the anti-rachitic molecule in cod liver oil, vitamin D.53
 In the past preterm infants were at risk for rickets because of an inadequate supply of calcium and phosphorus at the time of birth.
In the past preterm infants were at risk for rickets because of an inadequate supply of calcium and phosphorus at the time of birth.
Following feeding with unsupplemented breast milk, their phosphorus levels would fall over the first 2 weeks of life with a subsequent increase in alkaline phosphatase activity at 4-8 weeks, indicating vitamin D deficiency.54
 Since the introduction of breast milk fortifiers and preterm formulas, the incidence of rickets of prematurity has decreased dramatically. Rickets also occurs with greater frequency during puberty, when physical and behavioral changes modulate vitamin D availability. The pubertal growth spurt places greater demands for more calcium and phosphorus needed for bone growth, so increased levels of vitamin D are required. In settings where women are expected to cover their skin with the onset of puberty for cultural reasons, the scant opportunities for sun exposure increases the risk for vitamin D deficiency if dietary intake is insufficient to meet daily requirements.
Since the introduction of breast milk fortifiers and preterm formulas, the incidence of rickets of prematurity has decreased dramatically. Rickets also occurs with greater frequency during puberty, when physical and behavioral changes modulate vitamin D availability. The pubertal growth spurt places greater demands for more calcium and phosphorus needed for bone growth, so increased levels of vitamin D are required. In settings where women are expected to cover their skin with the onset of puberty for cultural reasons, the scant opportunities for sun exposure increases the risk for vitamin D deficiency if dietary intake is insufficient to meet daily requirements.
Vitamin D-deficient rickets continues to occur in modern times. Groups at risk for vitamin D deficiency include those with inadequate diet, malabsorption, and decreased exposure to sunlight. This includes the elderly or debilitated who have decreased sun exposure or decreased vitamin intake; patients on anticonvulsant therapy; those with malabsorption from gastrointestinal surgery, celiac disease, or pancreatic or biliary disease; those with chronic renal failure; dark-skinned individuals living in areas with poor sun exposure; and breast-fed babies exclusively breastfed without vitamin supplementation.
A recent resurgence in vitamin D-deficient rickets has prompted further evaluation of those at risk. A review of 166 cases of rickets in the United States between 1986 and 2003 showed that most cases presented between 4 and 54 months of age. Eighty-three percent were African-American or black and 96% were breastfed.55
These results emphasized that exclusively breastfed infants, especially those with dark skin tones, may require vitamin supplementation. In response to the continued increase in cases of vitamin D-deficient rickets, the American Academy of Pediatrics in 2003 outlined three populations who should be given supplemental vitamin D (200 IU): 1) all breastfed infants unless they take in 500 mL/day of fortified formula or milk; 2) all non-breastfed infants taking in less than 500 mL/day of fortified formula or milk; and 3) children and adolescents who do not obtain regular sun exposure, do not ingest at least 500 mL of fortified milk a day, or do not take a multivitamin with at least 200 IU vitamin D.56
Vitamin D-deficient rickets has been associated with congenital ichthyoses, such as lamellar ichthyosis,57–59 nonbullous ichthyosiform erythroderma,60 X-linked ichthyosis,58 and epidermolytic hyperkeratosis.61 Factors contributing to vitamin D deficiency include avoidance of sun exposure, excessive transepidermal calcium loss, defective vitamin D synthesis in affected skin, and decreased intestinal calcium absorption secondary to systemic retinoid therapy.
Given the movement to encourage sunscreen use, there has been concern regarding secondary vitamin D deficiency. Theoretically, regular use of the recommended amount of sunscreen can decrease 25-hydroxyvitamin D levels, but with real-life application of inadequate amounts of sunscreen and the tendency for increased sun exposure in individuals wearing sunscreen, there seems to be no significant impact on the incidence of vitamin D deficiency.62
At the same time, it appears that only limited sunlight exposure is necessary to produce adequate amounts of vitamin D3. For patients with Fitzpatrick skin type II, it has been calculated that only 5 minutes of summertime noon sun 2–3 times weekly provides adequate vitamin D production to satisfy physiologic requirements (see also Chapter 90).63,64
The classic manifestations of vitamin D-deficient rickets are skeletal (Box 130-7). Calcium and phosphorus deficiency leads to poor calcification of new bones, resulting in fraying and widening of the metaphysis. This can be seen at costochondral junctions of the anterior ribs, creating the well-known “rachitic rosary.” Poor calcification of the skull bones results in craniotabes, a softening of the skull bones giving them a ping-pong ball feel. As the bones become weaker, they cannot support the weight of the child and progressive lateral bowing of the lower extremities occurs. Other findings can include frontal bossing, widening of the wrists, scoliosis, hypotonia, fractures, dental defects, and rarely hypocalcemic seizures or tetany. Early radiographic signs of rickets include widening of the epiphyseal plate and blurring of the epiphyseal and metaphyseal junction. If the disease progresses, deformities at the growth plate develop, including cupping, splaying, formation of cortical spurs, and stippling. The bone cortices appear thinner and generalized osteopenia is noted.
A potentially fatal manifestation of vitamin D deficiency is a dilated cardiomyopathy. In a report of 16 British cases, three infants died and six additional infants were successfully resuscitated from cardiopulmonary arrest.65 Importantly, the cardiomyopathy is responsive to vitamin D supplementation and can result in complete resolution.65–67
Vitamin D-dependent rickets type II has also been associated with cutaneous features that are clinically indistinguishable from the syndrome of generalized atrichia associated with mutations in the hairless gene.68,69 Affected patients in both conditions are born with hair. However, within a few months after birth, scalp and body hair are lost with the exception of eyebrows and eyelashes. Small papules and cysts representing abnormal, rudimentary hair structures characteristically develop on the face and scalp. These cysts typically show disintegration of the lower two-thirds of the follicular unit. While the cutaneous features—notably the alopecia and cysts—are phenotypically and histologically identical, these are distinct clinical entities (Table 130-1).
Vitamin D-Resistant Rickets Type I | Generalized Atrichia |
|---|---|
Chromosome 12q14 | Chromosome 8p12 |
Mutations in vitamin D receptor (Zn finger) | Mutations in human hairless gene (Zn finger) |
End-organ unresponsiveness to Vitamin D | Defect in catagen remodeling |
Atrichia with papules and milia; + eyebrows/eyelashes | Atrichia with papules and milia; + eyebrows/eyelashes |
Alopecia by 1–3 months of age | Alopecia by 40 days to 4 months |
In addition to the clinical and radiological signs of rickets, laboratory examination may be helpful. Elevated alkaline phosphatase levels and low serum 25-hydroxyvitamin D levels are often useful laboratory indicators of vitamin D deficiency. In the early stages of rickets, parathyroid hormone levels increase to compensate, but this compensatory mechanism becomes inadequate if the deficiency continues.
The recommended daily value of vitamin D is 5–10 μg. Treatment includes oral vitamin D repletion with dihydroxyvitamin D in addition to a calcium-rich diet. Supplementation with 200–400 μg vitamin D per day until resolution of symptoms, about 2–3 months, is usually adequate.176 Additional therapy can include judicious sun exposure.
Two additional therapies can be used in cases of hepatic rickets, which is unresponsive to oral vitamin D supplementation because of decreased intraluminal bile salts. d-α-tocopheryl polyethylene glycol-1,000 succinate (TPGS), a water-soluble vitamin E that forms micelles at low concentrations, enhances vitamin D absorption and successfully treated eight pediatric cases of hepatic rickets. These patients maintained adequate levels of vitamin D while on continued TPGS and vitamin D supplementation, without elevation of vitamin E levels.70 Promoting cutaneous synthesis of vitamin D through ultraviolet radiation successfully treated two cases of hepatic rickets secondary to chronic cytomegalovirus hepatitis and to Alagille’s syndrome.71 Ultraviolet light therapy has also treated an Asian male with poor dietary intake of vitamin D.72
Vitamin E is rarely associated with deficiency or excess states of disease. Found in oils and shortenings, as well as various fortified grains, dark-green leafy vegetables, legumes, nuts, avocado, and small fishes such as herring and sardines.73 Because it is a fat-soluble vitamin, excessive intake may augment the effects of anticoagulant medications leading to purpura and propensity for hemorrhage.74 Deficiency states are rare. However, ataxia with isolated vitamin E deficiency (AVED) is a rare and severe spinocerebellar neurodegenerative disorder with autosomal recessive inheritance. Patients with mutations in the α-tocopherol transfer protein are unable to properly transfer α-tocopherol from lysosomes into lipoproteins that results in a predisposition to oxidative stress in affected cells.75–77
Vitamin K is a necessary cofactor in the carboxylation of glutamate residues on coagulation factors II, VII, IX, X, and proteins C and S. Dietary vitamin K, phylloquinone, is found in green, leafy vegetables, certain legumes, soybeans, cereals, and beef liver. Phylloquinone is actively transported in the distal small bowel. Approximately half of the body’s vitamin K is obtained though these dietary sources, and the other half is synthesized by gastrointestinal flora as menaquinones, which are passively absorbed in the distal small bowel and colon.
Vitamin K is derived from the German word “Koagulationsvitamin,” which literally translates to mean “clotting vitamin.” In the early-1900s, Henrik Dam of Denmark discovered an “antihemorrhagic factor” that reversed diet-induced bleeding disorders in chicks. In 1943, Edward Doisy and Henrik Dam were awarded the Nobel Prize in Physiology and Medicine for their separate work on isolating vitamin K.
Vitamin K deficiency leads to impaired coagulation and hemorrhage, which in neonates is referred to as hemorrhagic disease of the newborn (HDN). Neonates are particularly prone to vitamin K deficiency because of poor transplacental transfer, low dietary intake, and a sterile bowel. HDN is divided into early presentation and late presentation. The incidence of early HDN is 0.25%–1.7% and causes unexpected bleeding in the first week of life in an otherwise healthy neonate. It can present as ecchymoses, cephalohematomas or nasal, subgaleal, umbilical, intestinal, or intracranial hemorrhages. Late HDN is defined by the American Academy of Pediatrics as unexpected bleeding from severe vitamin K deficiency in 2–12 week old infants who are primarily breastfed and who received no or inadequate neonatal vitamin K prophylaxis.78 Vitamin K deficiency beyond the newborn period is rare, but may result from malabsorption, liver disease, inadequate dietary intake, or medications. Fat malabsorption occurs in conditions such as regional ileitis, topical sprue, celiac disease, cystic fibrosis, pancreatic insufficiency, and biliary obstruction. Antibiotic use can result in vitamin K deficiency by altering the populations of normal bowel flora. Coumarin interferes with vitamin K epoxide reductase, an enzyme important in the recycling of inactive vitamin K into its active form. Other medications that can interfere with vitamin K metabolism include anticonvulsants (phenytoin), rifampin, isoniazid, high-dose salicylates, cholestyramine, and cephalosporins.11,79 Vitamin K deficiency in older children and adults can present as purpura, ecchymoses, gingival bleeding, and gastrointestinal, genitourinary, and retroperitoneal hemorrhage.
Because vitamin K is a key cofactor in the coagulation pathway, deficiency of vitamin K typically manifests as elevations in both prothrombin time and activated partial thromboplastin time (PT and PTT). Serum levels of vitamin K can also be measured.
While des-γ-carboxyprothrombin (DCP), also known as the abnormal “protein induced by vitamin K absence” (PIVKA), can be a sensitive indicator for vitamin K deficiency, its presence has also been strongly linked to certain malignancies, in particular, hepatocellular carcinoma. It appears that hepatocellular carcinoma cells produce DCP directly rather than as a byproduct of low vitamin K levels, which may be normal in patients with hepatocellular carcinoma.80
Neonatal prophylaxis is traditionally with a single intramuscular dose of 0.5–1.0 mg vitamin K. There have been some studies regarding the use of oral vitamin K prophylaxis, but there is no definitive data on efficacy, safety, or bioavailability.78
Acute treatment is with fresh frozen plasma to replace deficient coagulation factors. Vitamin deficiency can also be treated with parenteral or intramuscular 5–10 mg vitamin K per day to correct severe deficiency.
|
Disorders of thiamine may have broad-ranging implications because thiamine is an essential coenzyme for three separate enzymes involved in NADPH synthesis, carbohydrate metabolism, and deoxyribose and ribose synthesis. Thiamine is used as a coenzyme for transketolase in the pentose phosphate pathway to produce NADPH. Thiamine pyrophosphate acts as a coenzyme in pyruvate dehydrogenase and α-ketoglutarate dehydrogenase, which are involved in oxidative decarboxylation reactions in the metabolism of carbohydrates and branched-chain amino acids.
Thiamine is obtained from whole grains, enriched bread products, dried peas and beans, potatoes, and fish. Polished rice eliminates the thiamine-containing husk and predisposes to thiamine deficiency.
Disorders of thiamine excess are extremely rare. Most arise as a result of intravenous administration for suspected thiamine deficiency in the context of chronic alcoholism. Local irritation at the site of intravenous administration, generalized pruritus, and anaphylactic or anaphylactoid reactions have been described.81 Neurotoxicity can occur in experimental settings when thiamine is administered directly into the central nervous system.82 In general, thiamine excess states are extremely rare in humans.
Beriberi refers to a thiamine deficiency state. The word is derived from Sinhalese meaning “extreme weakness.” The symptoms of beriberi have been recognized in East Asian countries for thousands of years because polished white rice is a dietary staple. The Japanese navy observed in the 1890s that beriberi could be eradicated by adding meat, fish, and vegetables to the diet.83 Beriberi became an epidemic in the Dutch East Indies in the late 1800s. Christiaan Eijkman was part of the medical team stationed in the Dutch East Indies to study beriberi. In 1929, Eijkman was awarded the Nobel Prize in Physiology and Medicine for his work starting in 1886 studying the effect of polished rice and unpolished rice on the incidence of beriberi in chickens. Through a series of detailed experiments on populations of chickens fed various diets and injected with various bacteria, he concluded that there was a direct correlation between diet and beriberi, but like many, had initially misattributed the cause to a nonexistent infectious agent in polished rice. In 1926, Barend Coenraad Petrus Jansen and William Frederick Donath successfully isolated thiamine from rice polishings, and Robert Williams was able to synthesize thiamine in the 1930s.84
Thiamine deficiency in the United States is now rare. Predisposing factors for pediatric thiamine deficiency include unsupplemented parenteral nutrition, breastfed infants of thiamine-deficient mothers, congestive heart failure,85 and severe malnutrition. Early signs include irritability, apathy, restlessness, and vomiting. As the disease progresses, neurologic signs of Wernicke’s encephalopathy may develop, such as ophthalmoplegia, ataxia, nystagmus, and characteristic laryngeal nerve paralysis resulting in aphonia, which is a classic manifestation of infantile beriberi. Other symptoms include congestive heart failure, tachycardia, dyspnea, and cyanosis. In 2003, a series of infants presenting with ophthalmoplegia as a manifestation of Wernicke’s encephalopathy was reported in Israel as a result of a thiamine-deficient infant soy formula. In all these cases, a prodromal illness was observed. Early symptoms included vomiting, anorexia, diarrhea, lethargy, irritability, and developmental delay. Upbeat nystagmus and ophthalmoplegia were the primary neurological signs. Following treatment, those with early disease had complete recovery, but those with severe disease had residual neurological complications.86
Adult beriberi has been categorized into dry and wet forms. Dry beriberi describes a symmetric distal peripheral neuropathy involving both sensory and motor systems. Wet beriberi includes neuropathy and signs of cardiac involvement, including cardiomegaly, cardiomyopathy, congestive heart failure, peripheral edema, and tachycardia. Rarely, wet beriberi can be associated with pulmonary hypertension that is reversible after thiamine supplementation.87 A red, burning tongue and peripheral edema have also been observed with wet beriberi.
Diagnosis of thiamine deficiency is made by measurement of erythrocyte thiamine transketolase or blood thiamine concentration. The more reliable measure is erythrocyte thiamine transketolase before and after thiamine pyrophosphate stimulation, represented as a percentage of thiamine pyrophosphate effect (TPPE). Normal values are up to 15%.
Because thiamine is a cofactor in a variety of metabolic pathways, daily thiamine requirements are calculated from an individual’s ideal total caloric intake, with current recommendations indicating 0.5 mg per 1,000 kcal.
Treatment of thiamine deficiency can be via intravenous, intramuscular, or oral routes of administration. Usually, treatment for beriberi is initiated with intravenous or intramuscular thiamine of 50–100 mg per day for 7–14 days, then oral supplementation is provided until full recovery is documented.
Riboflavin was discovered in 1879 as a fluorescent yellow–green substance found in milk. Its chemical structure was later determined in 1933. Riboflavin is used in two coenzymes, (1) flavin mononucleotide (FMN) and (2) flavin-adenine dinucleotide (FAD), both of which are involved in oxidation-reduction reactions in cellular respiration and oxidative phosphorylation. These two enzymes are also involved in pyridoxine (vitamin B6) metabolism. Recent studies have suggested that riboflavin deficiency may contribute to increase plasma homocysteine levels, impaired handling of iron, and night blindness.88
Riboflavin is typically obtained through dairy products, nuts, meat, eggs, whole grain and enriched bread products, fatty fish, and green leafy vegetables. A small amount of dietary riboflavin is present as free riboflavin; most are found as FAD or FMN. Dietary FAD and FMN are hydrolyzed to riboflavin by brush border membranes or enterocytes. Free riboflavin in the intestinal lumen is then taken up by active transport in the proximal small bowel.
Deficiency states can be caused by decreased intake, inadequate absorption, and phototherapy. Alcoholics, elderly, and adolescents are groups at risk for riboflavin deficiency secondary to poor nutritional intake. Malabsorption after gastric bypass surgery can also predispose individuals to riboflavin deficiency.89 In areas of India, China, and Iran, riboflavin deficiency is endemic because of their dependence on an unenriched cereal diet. Infants of riboflavin-deficient mothers are also at risk of deficiency because breast milk concentrations of the vitamin are proportional to maternal concentrations. Once weaned from the breast, these infants are at additional risk if they are not transitioned to milk. When confounded by PEM, riboflavin deficiency may be worsened because the usual renal compensatory mechanism of increased riboflavin absorption is impaired in this setting. Visible light phototherapy for jaundiced neonates causes photodecomposition of riboflavin.90 Certain drugs also affect riboflavin levels through effects on absorption or metabolic inhibition. Chlorpromazine and other tricyclic drugs inhibit transport of riboflavin in the gastrointestinal tract predisposing to deficiency states.91 Borate displaces riboflavin from binding sites, increases urinary riboflavin excretion, and inhibits riboflavin-dependent enzymes contributing to riboflavin deficiency.92
Signs of acute riboflavin deficiency include a deep red erythema, epidermal necrolysis, and mucositis. The severity of symptoms depends on the severity of deficiency93 (Box 130-8).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


