Cutaneous Changes in Errors of Amino Acid Metabolism: Introduction
Inborn errors of amino acid metabolism are inherited defects of enzymes that result in important disturbance in amino acid biology.1 Most of them are autosomal recessive disorders. Amino acids are the molecular units that make up proteins, and all proteins are various combinations of 20 specific naturally occurring amino acids.
Diseases of amino acid metabolism may lead to multiorgan disorders with signs and symptoms of mental retardation and dysfunction in numerous additional organs, including skin and hair.2 With more sophisticated diagnostic techniques, metabolic diseases have gained increasing importance, and numerous rare entities have been identified, including hyperphenylalaninemias, hypertyrosinemias, disorders of histidine metabolism, disorders of proline and hydroxyproline metabolism, hyperornithinemias, dysfunctions of urea cycle enzymes, disorders of lysine metabolism, disorders of branched-chain amino acid and keto acid metabolism, disorders of transsulfuration with homocystinuria and homocystinemia, sarcosinemia, and nonketotic hyperglycinemia.3
Several diseases have important cutaneous manifestations that might be a clue to diagnosis (Table 131-1). An example of such a key manifestation representing errors of amino acid metabolism is acrodermatitis acidemica. These patients have characteristic cutaneous lesions that are similar to those in acrodermatitis enteropathica (see Chapter 130). Psoriasiform dermatitis on the trunk and extremities may occur together with diffuse alopecia and brittle hair.4 In general, acral dermatitis with perioral erythema and desquamation is associated with poor feeding, lethargy, hypotonia, vomiting, and dehydration. Patients often have progressive neurologic symptoms. Patients with branched-chain aminoacidopathies may have methylmalonic acidemia, propionic acidemia, glutaric acidemia, biotinidase deficiency, multiple carboxylase deficiency, or maple syrup urine disease, all of which are sometimes accompanied by cutaneous problems.
Disease | Skin Manifestations | Deficient Enzyme/Pathophysiology (Mode of Inheritance) |
|---|---|---|
Alkaptonuria | Ochronosis Black cerumen, eccrine, and apocrine secretions | Homogentisic acid oxidase (AR) |
Argininosuccinic aciduria | Trichorrhexis nodosa Rough skin | Argininosuccinase (AR) |
Aspartylglycosaminuria | Thick skin Coarsening of face Sagging skinfolds Increased acne Photosensitivity | Aspartylglycosamidase (AR) |
Biotinidase deficiency | Erythematous rash Alopecia Oral candidiasis Seborrheic dermatitis Glossitis | Biotinidase (AR) |
Citrullinemia | Light, short hair with interrupted cuticle | Argininosuccinate synthetase (AR) |
Hartnup disease | Pellagra-like lesions Photosensitivity | Defect of neutral amino acid transport in renal and intestinal brush border (AR); increases excretion of tryptophan metabolites |
Histidinemia | Light-colored hair and eyes | Histidase (AR) |
Holocarboxylase synthetase deficiency | Seborrheic dermatitis Alopecia | Holocarboxylase synthetase (AR) |
Homocystinuria | Fine, sparse, friable hair Thin skin Livedo reticularis Malar flush Vascular occlusions Marfanoid habitus | Cystathionine β-synthetase (AR) |
Hydroxykynureninuria | Chronic stomatitis Ulcerated gums, gingivitis Photosensitivity | Kynureninase (AR) |
Hyperprolinemia | Ichthyosis | Proline oxidase (AR) |
Iminodipeptiduria (prolidase deficiency) | Chronic skin ulcers Recurrent infections | Prolidase (AR) |
Isovaleric acidemia | Odor of sweaty feet, alopecia | Isovaleryl-coenzyme A dehydrogenase (AR) |
Methionine malabsorption syndrome | White hair Dried celery or oasthouse odor Edema | Defective methionine transport |
Phenylketonuria | Hypopigmentation Atopic dermatitis Scleroderma | Phenylalanine hydroxylase (AR) Dihydropterine reductase Defective dihydropterine synthesis |
Tyrosinemia II | Painful, acral erosions Hyperkeratosis Corneal/conjunctival plaques and erosions | Tyrosine aminotransferase (AR) |
Xanthurenic aciduria | Urticaria | Kynureninase (AD) |
In general, all patients with acrodermatitis acidemica have absolute missing enzyme activity of their autosomal recessive mutated gene. Diagnosis is confirmed by documentation of increased amounts of amino acids in the urine. Exact diagnosis is the first step for adequate treatment of these diseases.
Hartnup disease results from a deficiency of neutral amino acid transport within the kidneys and small intestine and leads to pellagra-like features in a photodistribution (see Chapter 130). Neurologic deficiencies and reduced mental skills are often associated. The treatment of choice is nicotinamide.
Tyrosinemias
|
The genetic tyrosinemias are characterized by the accumulation of tyrosine in body fluids and tissue.5,6 Tyrosine is a semiessential amino acid, derived from the liberation of tyrosine from hydrolysis of dietary or tissue protein, or from the hydroxylation of the essential amino acid phenylalanine, and is the starting point for the synthesis of catecholamines, thyroid hormones, and melanogenesis. There are three types of tyrosinemia. The skin is not involved in tyrosinemia types I and III, but it is involved in tyrosinemia II, which is also called the oculocutaneous tyrosinemia and Richner–Hanhart syndrome [Online Mendelian Inheritance in Man (OMIM) #276600]. Tyrosinemia II is a rare distinctive clinical symptom complex involving the eyes, skin, and central nervous system and is potentially treatable. Tyrosine levels are elevated because of a deficiency of hepatic tyrosine aminotransferase (TAT).
Approximately 100 patients with this clinical syndrome have been reported. Goldsmith and Reed were the first to correlate the oculocutaneous syndrome with an underlying tyrosinemia.7 All patients had tyrosinemia, phenolic aciduria, and inflammatory skin and eye lesions. The sexes are affected equally, and the disease is worldwide in distribution. Transmission of tyrosinemia II is autosomal recessive. Consanguinity has been documented, and occurrence in several siblings within a family is well known (pseudodominant pattern). Heterozygotes are unaffected clinically and demonstrate no biochemical alterations of tyrosine or its metabolites in blood or urine.
Patients have hyperkeratotic yellowish skin lesions limited to the palms and soles, which in 80% of cases occur on the peripheral pressure-bearing areas.5,7 Lesions usually begin during the first year of life. The skin lesions are painful, nonpruritic, and frequently associated with hyperhidrosis (Fig. 131-1). Diffuse plantar keratoderma and self-mutilation have been observed in individuals with tyrosinemia II.8 Leukokeratosis of the tongue has been reported. Bullous lesions may occur and progress rapidly to erosions; these then become crusted and hyperkeratotic.9 The fingertips and hypothenar eminences are commonly involved. Rare patients have hyperpigmentation and/or hyperkeratotic subungual lesions, and in older persons hyperkeratosis in the elbows, knees, and ankles may appear.2
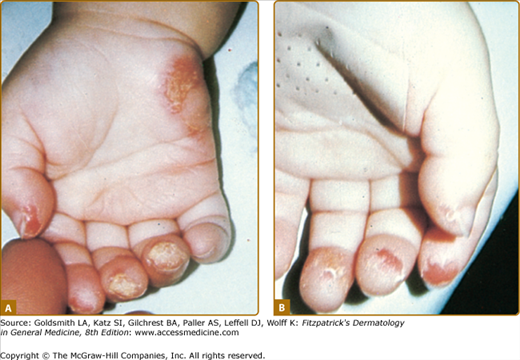
The most important manifestations of oculocutaneous tyrosinemia are those involving the eye, because they can lead to permanent visual impairment.10 Eye lesions occur weeks to months before the skin lesions. Ophthalmologic symptoms start as early as the first day of life and as late as 38 years of age. Tearing, redness, pain, and photophobia are early signs and symptoms; late findings include corneal clouding and central or paracentral opacities, which are initially intraepithelial and can progress to superficial or deep pseudodendritic keratitis (Fig. 131-2). Slit-lamp examination may reveal some degree of corneal ulceration, and occasionally birefringent crystals of tyrosine may be observed. Neovascularization is prominent. The eye disease occurs in 75% of affected patients and may lead to scarring, nystagmus, and exodeviation. Ocular symptoms may show spontaneous remission and recurrences and may occur independently of other manifestations. Topical therapy is ineffective, and herpes simplex virus and bacterial cultures consistently yield negative results.
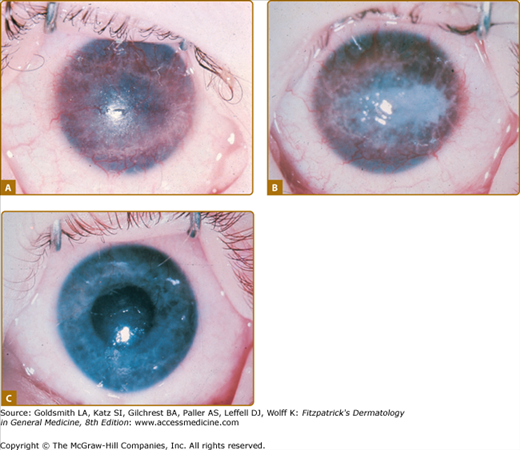
Mental retardation of varying degrees is reported in half the cases, as is normal mental development. Hyperactivity has been observed in several affected children, as has abnormal language development. Untreated tyrosinemia II during pregnancy may be associated with mental retardation or seizures in the children resulting from that pregnancy.11
In establishing the diagnosis in a child with tyrosinemia II, laboratory verification of plasma amino acid and urine organic acid levels is mandatory. The blood and urine tyrosine levels of affected patients are markedly elevated. Levels of other amino acids are not increased. Urinary tyrosine metabolite levels are elevated; these include p-hydroxyphenylpyruvic acid, p-hydroxyphenyllactic acid, p-hydroxyphenylacetic acid, and N-acetyltyrosine (Fig. 131-3). All these metabolic effects are consequences of the deficiency of hepatic TAT. A fluorometric procedure exists as well as a tandem mass spectrometry, which is used for neonatal screening. Recently, an asymptomatic newborn was detected to have Richner–Hanhart syndrome on the third day of life as a result of the newborn screening. Interestingly the 8-year-old brother with persistent plantar hyperkeratotic plaques of the soles of yet unknown origin was subsequently identified to be also affected tyrosinemia type II.12
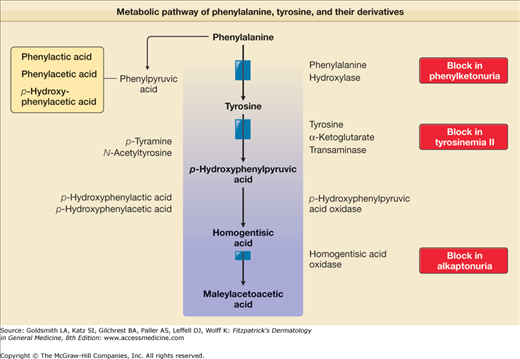
TAT is a pyridoxal phosphate-dependent cytoplasmic enzyme that transaminates tyrosine, forming p-hydroxyphenylpyruvate (PHPPA). The human TAT gene contains 12 exons and transcribes a 2.75-kb messenger RNA (mRNA) that codes for a 454-amino acid protein.5 The liver is the richest source of TAT; this specific TAT is not present in skin. In tyrosinemia II, the liver biopsy specimen shows little or no soluble TAT, although there is normal or slightly increased mitochondrial tyrosine (aspartate) transaminase activity.12,13 Mitochondrial aspartate aminotransferase using tyrosine as a substrate produces increased amounts of PHPPA from the increased amounts of tyrosine available in tyrosinemia II. Because mitochondria do not have PHPPA oxidase activity, PHPPA and its metabolic products increase and appear in the urine, which creates the unusual situation in which metabolites are increased both proximally and distally to the defective enzyme.
Routine histopathologic analysis of the skin is not diagnostic; it shows hyperkeratosis, parakeratosis, and acanthosis.9 In some cases, electron microscopy studies have shown lipid droplets in the cornified layer, increases in tonofibrils and keratohyalin, very tightly packed microtubular and microfibrillar masses, and minute tyrosine crystals in keratinocytes and Merkel cells.14
Mutations occur in the human TAT gene, which results in high levels of tyrosine. TAT maps to the q22-q24 region of chromosome 16 and has been sequenced.5 Multiple point mutations scattered throughout the TAT gene have been associated with tyrosinemia II.15
Interestingly, in mink with the disease TAT unlinked gene regulatory mutation rather than TAT deficiency was thought to be the cause of tyrosinemia.16
PATHOPHYSIOLOGY OF THE SKIN AND EYE LESIONS IN TYROSINEMIA II
 Rats fed a 12 percent protein diet with 0.5 percent to 2.0 percent tyrosine developed a syndrome resembling tyrosinemia II, with weight loss, a shortened life span, keratitis, conjunctivitis, alopecia, cheilitis, and inflammatory toe changes.17 The high-tyrosine syndrome in rats was ameliorated by increasing the protein content of the diet or by adding threonine or thiouracil or TAT inducers.
Rats fed a 12 percent protein diet with 0.5 percent to 2.0 percent tyrosine developed a syndrome resembling tyrosinemia II, with weight loss, a shortened life span, keratitis, conjunctivitis, alopecia, cheilitis, and inflammatory toe changes.17 The high-tyrosine syndrome in rats was ameliorated by increasing the protein content of the diet or by adding threonine or thiouracil or TAT inducers.
In vitro studies showed that tyrosine crystals can cause release of lysosomal enzymes and as yet uncharacterized heat-labile chemotactic factors.18,19 Crystal-induced inflammation has been studied in model systems using calcium pyrophosphate dihydrate and monosodium urate monohydrate.
(See Box 131-1). Corneodermatoosseous syndrome (OMIM #122440), an autosomal dominant symptom complex of volar keratosis and keratitis that is clinically similar to tyrosinemia II, has been described in one family.20 The lesions were more extensive than those in the classic Richner–Hanhart syndrome. In addition, brachydactyly with short distal phalanges, short stature, and soft teeth may occur.
Main Features
|
Consider
|
Always Rule Out
|
With consumption of a low-tyrosine, low-phenylalanine diet (Mead Johnson, Tyromex-Ross), there is a rapid decrease in tyrosine to normal levels (30–90 μM). Skin and eye lesions cleared within days and weeks in all individuals treated with the diet7,12 (see Fig. 131-2C; Fig. 131-4; Box 131-2).
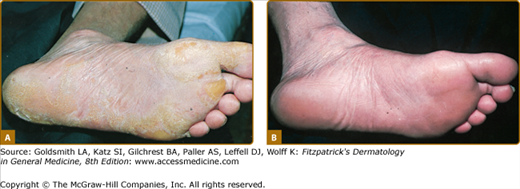
Some patients have responded objectively to etretinate, although plasma tyrosine levels remained unchanged.21 In none of the patients studied has there been a response to cortisone acetate, ascorbic acid, pyridoxine, or folic acid, which are cofactors or known inducers of TAT and PHPPA oxidase. Surgical treatment is recommended for palmar lesions but not for the plantar form.
Because the consequences of tyrosinemia II are serious and a safe treatment is available, a patient presenting with any atypical bullous or hyperkeratotic disease on the palms and soles in the first months of life should be screened for tyrosine and its metabolites; screening tests are available in most hospital laboratories.22 Amino acid analysis is necessary to confirm the diagnosis and follow the response to diet therapy. Dietary control in pregnancy should be recommended. A patient with tyrosinemia II was reported who had undergone two untreated pregnancies. During the pregnancies, plasma tyrosine level was raised, and the infants were retarded. On the other hand, several normal children of mothers with the disease have been observed.23
Phenylketonuria
Phenylketonuria (phenylpyruvic oligophrenia, PKU) is an autosomal recessive disorder of aromatic amino acid metabolism (OMIM #261600) in which phenylalanine cannot be converted to tyrosine. Its discovery over 70 years ago has established the link between metabolic disease and intellectual impairment, and led to the development of neonatal screening programs across the globe. In addition, it demonstrated how effective treatment could lead to a near normal outcome for affected individuals. PKU is caused by a deficiency of hepatic phenylalanine hydroxylase or cofactors for phenylalanine hydroxylase. PAH deficiency seems to be a protein-misfolding disease in which global conformational changes hinder molecular motions essential for physiological enzyme function.24 There are additional causes that may lead to hyperphenylalaninemias, and eight subtypes have been described. Maternal PKU may lead to hyperphenylalaninemia. Unless the mother follows a special diet, the child is at risk to develop the typical signs and symptoms of PKU. Defects of biopterin metabolism, prematurity, liver disease, use of medications such as methotrexate, and high-protein diets may produce findings similar to those in PKU. Genotype-based prediction and classification of the biochemical phenotype is now possible in the majority of newborns with hyperphenylalaninemia.24
|
The prevalence of PKU varies markedly in various countries. The highest incidence of PKU is found in Ireland and Scotland (1 in 4,000), whereas in Finland only 1 in 40,000 births are affected. The overall frequency among northern Europeans was calculated to be approximately 1 in 10,000 and in the United States classic PKU was estimated to occur in approximately 1 in 11,000 births in the white population. Among blacks the frequency was estimated to be only approximately 1 in 50,000.25
The clinical hallmarks of PKU are mental retardation, diffuse hypopigmentation, seizures, eczematous dermatitis, and photosensitivity.26 Newborns appear to be normal and develop manifestations of psychomotor alterations between 4 and 24 months of age. Early symptoms include heavy vomiting. The most important defects are marked mental retardation with irritability, hyperactivity, self-destructive behavior, hypertonicity, and seizures. Peculiar gait and increased deep tendon reflexes are noted. Microcephaly, brain calcification, and cataracts are observed quite commonly. Typically, a mousy odor induced by phenylacetic acid is noted in urine and sweat.
Early infantile eczema, indistinguishable from atopic dermatitis, or seborrheic dermatitis may be one of the first signs of PKU and occurs in 20%–50% of affected infants during the first year of life. Later on, the incidence may be even higher. Dramatic clearing of eczematous skin lesions with a low-phenylalanine diet has been reported in some patients, and a potential role for altered biotin recycling is suggested.27 A further skin clue in PKU is pigment dilution, with impressive pale pigmentation, blond hair, and blue eyes seen in 90% of patients. The pigment dilution and hair loss is reversible with phenylalanine restriction.28 The color changes may be especially striking in the rare black or Japanese patient with PKU, whose eye or hair color may be very different from that of other members of the ethnic group. The increased levels of phenylalanine and its oxidation products (phenylpyruvic acid, o-hydroxyphenylacetic acid, phenylacetic acid) inhibit the enzyme tyrosinase and therefore reduce melanization.28
Patients develop moderate photosensitivity. Edematous scleroderma-like induration of the skin of the extremities, which spares the hands and feet, is characteristic.29 Plaque, guttate or generalized morphea, and atrophoderma of Pasini and Pierini (see Chapter 64) as well as lichen sclerosus et atrophicus (see Chapter 65) have been observed in PKU.30
Diagnosis of PKU is made by documenting elevated levels of phenylalanine in the serum (20 mg/dL or higher, 10–50 times the normal level). Plasma tyrosine levels may be normal or elevated. In addition, elevated urinary phenylpyruvic acid levels can be documented. Additional laboratory abnormalities include phenylalanine hydroxylase deficiency, phenylpyruvic acidemia, and increased urinary levels of o-hydroxyphenylacetic acid and phenylacetylglutamine. Increased urinary levels of phenylpyruvic acid can be detected by a typical green coloration of urine after addition of 10% ferric chloride. In numerous countries, PKU screening is performed in the neonatal screening program either by the semiquantitative bacterial inhibition assay method in which several drops of capillary blood may show an elevated level of phenylalanine in a microbiologic procedure (Guthrie test) or by a quantitative chemical reaction method (Quantase test).31
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


