The complex and fascinating spectrum of inflammatory skin disease, and the comprehension of it, is ever expanding and evolving. During the first decade of the 21st century, numerous advances in the understanding of inflammatory disease mechanisms have occurred, particularly in psoriasis and atopic dermatitis. Continuation of this trend will assure a future in which molecular tests for biomarkers of immediate clinical relevance are used in routine patient care, not only for diagnosis but also for prognosis and management. This article focuses on selected recent or noteworthy developments that are clinically relevant for the histologic diagnosis of inflammatory skin diseases.
- •
Clinical correlation maximizes diagnostic accuracy in the histologic interpretation of inflammatory skin disease.
- •
Variations from the classic histopathologic features are increasingly recognized. Examples include sarcoid, Sweet syndrome, and paraneoplastic pemphigus.
- •
The clinical differential diagnosis for nonspecific histologic reaction patterns is increasingly recognized. Examples include interstitial granulomatous infiltrates, flame figures, and dermal hypersensitivity.
During the first decade of the 21st century, numerous advances in the understanding of inflammatory disease mechanisms have occurred, particularly in psoriasis and atopic dermatitis. Continuation of this trend will assure a future in which molecular tests for biomarkers of immediate clinical relevance are used in routine patient care, not only for diagnosis but also for prognosis and management.
This article is organized from the perspective of one approaching a new case at the microscope and follows the traditional pattern-based approach used in many dermatopathology textbooks and in the authors’ institution’s training program ( www.ucdermpath.org/detail/TrainingProgUCD.htm ). However, out of necessity, the authors assume that the reader possesses baseline knowledge of the basic clinical and histopathologic features of the most common disorders. The emphasis is focused on selected recent or noteworthy developments that are clinically relevant for the histologic diagnosis of inflammatory skin diseases.
Spongiotic dermatitis
Spongiotic dermatitis remains one of the more commonly biopsied inflammatory dermatoses, yet distinction among nummular, atopic, irritant contact, allergic contact, dyshidrotic dermatitis, id reactions, spongiotic drug reactions, and rare paraneoplastic reactions continues to require clinical correlation.
Spongiotic Dermatitis Versus Mycosis Fungoides
The differential diagnosis of early patch-stage mycosis fungoides versus spongiotic dermatitis remains a challenge, clinically and histologically. Clinical correlation reveals that some of these cases may be classified as lymphomatoid drug reactions or lymphomatoid contact dermatitis, with a range of implicated allergens, including nickel, antioxidants, cosmetic-grade preservatives in baby wipes, and even teakwood toilet seats. In contrast to the epidermotropism that is a hallmark of mycosis fungoides, lymphomatoid reactions tend to exhibit appearances that may be characterized more descriptively as exocytosis, characterized by greater spongiosis (even if mild), mild or absent cytologic atypia, and a more randomly scattered pattern of lymphocytes throughout all levels of the epidermis (the epidermotropic lymphocytes in mycosis fungoides tend to collect in the basilar epidermis). When additional diagnostic analysis is required, demonstration of a clonal T-cell receptor (TCR) gene rearrangement provides additional support for lymphoma. However, clonality has been documented in some cases of nonneoplastic disorders (spongiotic dermatitis, pemphigoid, lichen planus, lichen sclerosus, psoriasis, pityriasis lichenoides, chronic cutaneous lupus erythematosus, and lymphomatoid drug reactions). Demonstrating the presence of an identical clone in two specimens (“dual TCR”) or more offers greater diagnostic specificity in support of lymphoma.
Prurigo Pigmentosa (Nagashima’s Disease)
Reports of this rare but clinically distinctive disorder originated in Japan, with rare subsequent reports from America and Europe. Prurigo pigmentosa presents as highly pruritic inflamed papules that coalesce into a reticulate pattern on the trunk and neck, perhaps appearing clinically as an inflammatory variant of confluent and reticulated papillomatosis. Histopathologic features have been regarded as nonspecific, but early lesions show combined spongiotic and interface features with neutrophilic spongiosis, necrotic keratinocytes, and papillary dermal edema ( Fig. 1 ). Established lesions may exhibit a patchy lichenoid mixed infiltrate with neutrophils and eosinophils, nuclear debris “dust,” reticular alteration and necrosis of the epidermis, and neutrophilic crust. Rare reports have associated Helicobacter pylori infection with prurigo pigmentosa, including a recent report in which H pylori was immunohistochemically confirmed within the stratum corneum of prurigo pigmentosa.
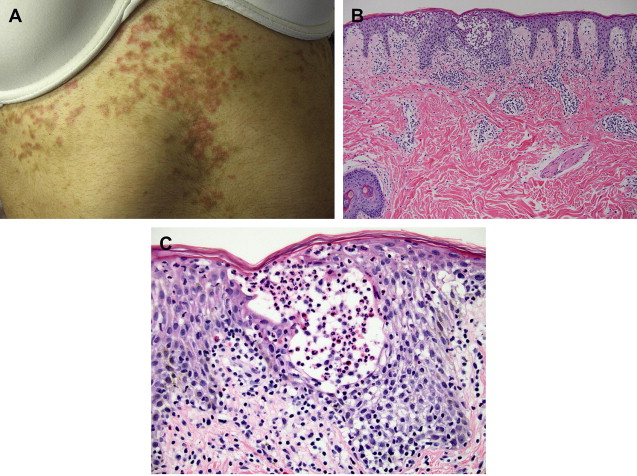
Psoriasiform dermatitis
The differential diagnosis for psoriasiform dermatitis typically includes psoriasis, psoriasiform drug reactions, chronic spongiotic/eczematous dermatitis, lichen simplex chronicus, and pityriasis rubra pilaris. Secondary syphilis and patch/plaque mycosis fungoides tend to have a superimposed lichenoid infiltrate, creating a psoriasiform lichenoid pattern.
Deficiency Dermatitis
Necrolytic migratory erythema (glucagonoma syndrome) remains a notorious mimic of psoriasis and should be considered in the appropriate clinical setting (hyperglycemia, anemia, glossitis) for any adult with an unusually eroded or refractory psoriasiform dermatitis ( Fig. 2 ). Compared with classic psoriasis, necrolytic migratory erythema exhibits confluent parakeratosis, hypogranulosis with pallor of the superficial epidermis, and eventual necrolysis (a term that encompasses vacuolization, ballooning degeneration, and subsequent confluent necrosis of keratinocytes within the superficial stratum spinosum and stratum granulosum). Nonspecific findings may also be seen, including mild spongiosis, single necrotic keratinocytes, and eosinophils; these features may be present to a limited extent in psoriasis but are nevertheless not classic for psoriasis. Necrolytic acral erythema exhibits similar histopathologic features to other forms of deficiency dermatitis but is confined to acral skin and is highly associated with hepatitis C virus (HCV) infection. Although HCV-associated leukocytoclastic vasculitis is well recognized, necrolytic acral erythema seems to develop in a different subset of patients who are HCV-positive, because vasculitis and necrolytic acral erythema have not been documented simultaneously in the same patient to date. Overlapping features of necrolytic acral erythema and necrolytic migratory erythema have been described, suggesting a common pathogenesis. The differential diagnosis for deficiency dermatitis also includes other nutritional deficiency states, the best characterized being zinc transporter protein (ZIP4/SLC39A4) mutation–associated zinc deficiency (acrodermatitis enteropathica, OMIM 201100), acquired zinc deficiency, and vitamin B 3 (niacin) deficiency (pellagra). Necrolytic acral erythema is often associated with hypozincemia and responds to zinc supplementation.
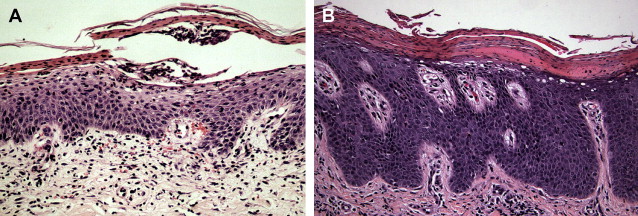
Pityriasis Rubra Pilaris
Biopsies from patients with pityriasis rubra pilaris (PRP) are characterized by psoriasiform hyperplasia, classic “checkerboard” parakeratosis, and sparse superficial perivascular lymphocytes. Comprehensive reviews of histopathologic features in PRP have confirmed additional features that may assist in the distinction between PRP and psoriasis, including the presence in PRP of hypergranulosis, acantholysis, and areas of atrophy amidst a background of psoriasiform hyperplasia, and eosinophils and denser lichenoid infiltrates in some cases. One case exhibited superficial acantholysis resembling pemphigus foliaceus.
Actinic Prurigo
Actinic prurigo (hereditary polymorphous light eruption, Hutchinson summer prurigo) shares many features with polymorphous light eruption (PMLE). Some classifications include actinic prurigo as a severe variant of PMLE, although consistent clinical and histologic differences have been documented that permit separation of actinic prurigo as a distinct disorder. Actinic prurigo usually arises in childhood and is more common in women. Strong HLA-associated susceptibility has been documented. Most cases in the United States have been reported in native North Americans, but actinic prurigo also affects Mestizos (mixed Native Indian and European) in Central and South America and has also been reported in the United Kingdom. As a clinically polymorphous eruption, actinic prurigo exhibits variable histopathologic features. Characteristic histopathologic features include psoriasiform epidermal hyperplasia, papillary dermal edema, and perivascular dermatitis (often dense, lichenoid, and/or deep) with eosinophils. More variable findings include solar elastosis, orthokeratosis or parakeratosis, spongiosis, vacuolar interface changes, and plasma cells. Reactive lymphoid follicles are a variable but distinctive finding. Thus, although it is expected that some cases of actinic prurigo will be clinically and histologically indistinguishable from PMLE, the presence of psoriasiform hyperplasia, eosinophils, and lymphoid follicles favors actinic prurigo ( Fig. 3 ). Specific clinical features of actinic prurigo include conjunctivitis and cheilitis, and the typical ethnicity/ancestry.
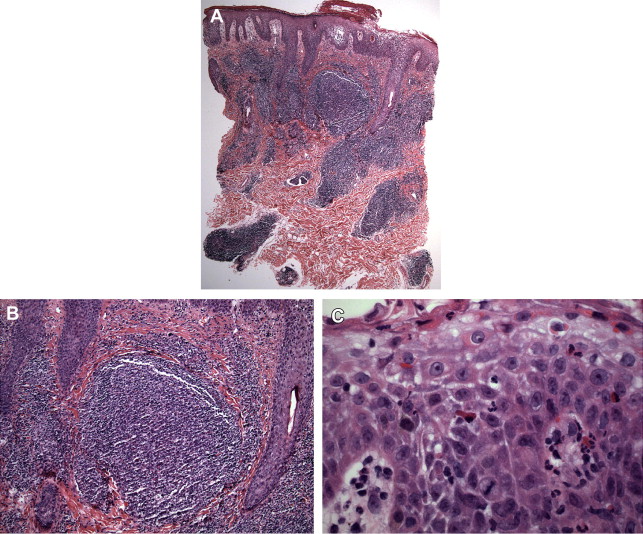
Psoriasiform dermatitis
The differential diagnosis for psoriasiform dermatitis typically includes psoriasis, psoriasiform drug reactions, chronic spongiotic/eczematous dermatitis, lichen simplex chronicus, and pityriasis rubra pilaris. Secondary syphilis and patch/plaque mycosis fungoides tend to have a superimposed lichenoid infiltrate, creating a psoriasiform lichenoid pattern.
Deficiency Dermatitis
Necrolytic migratory erythema (glucagonoma syndrome) remains a notorious mimic of psoriasis and should be considered in the appropriate clinical setting (hyperglycemia, anemia, glossitis) for any adult with an unusually eroded or refractory psoriasiform dermatitis ( Fig. 2 ). Compared with classic psoriasis, necrolytic migratory erythema exhibits confluent parakeratosis, hypogranulosis with pallor of the superficial epidermis, and eventual necrolysis (a term that encompasses vacuolization, ballooning degeneration, and subsequent confluent necrosis of keratinocytes within the superficial stratum spinosum and stratum granulosum). Nonspecific findings may also be seen, including mild spongiosis, single necrotic keratinocytes, and eosinophils; these features may be present to a limited extent in psoriasis but are nevertheless not classic for psoriasis. Necrolytic acral erythema exhibits similar histopathologic features to other forms of deficiency dermatitis but is confined to acral skin and is highly associated with hepatitis C virus (HCV) infection. Although HCV-associated leukocytoclastic vasculitis is well recognized, necrolytic acral erythema seems to develop in a different subset of patients who are HCV-positive, because vasculitis and necrolytic acral erythema have not been documented simultaneously in the same patient to date. Overlapping features of necrolytic acral erythema and necrolytic migratory erythema have been described, suggesting a common pathogenesis. The differential diagnosis for deficiency dermatitis also includes other nutritional deficiency states, the best characterized being zinc transporter protein (ZIP4/SLC39A4) mutation–associated zinc deficiency (acrodermatitis enteropathica, OMIM 201100), acquired zinc deficiency, and vitamin B 3 (niacin) deficiency (pellagra). Necrolytic acral erythema is often associated with hypozincemia and responds to zinc supplementation.
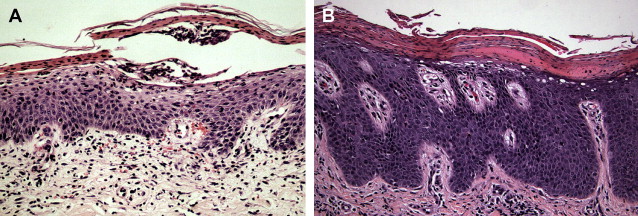
Pityriasis Rubra Pilaris
Biopsies from patients with pityriasis rubra pilaris (PRP) are characterized by psoriasiform hyperplasia, classic “checkerboard” parakeratosis, and sparse superficial perivascular lymphocytes. Comprehensive reviews of histopathologic features in PRP have confirmed additional features that may assist in the distinction between PRP and psoriasis, including the presence in PRP of hypergranulosis, acantholysis, and areas of atrophy amidst a background of psoriasiform hyperplasia, and eosinophils and denser lichenoid infiltrates in some cases. One case exhibited superficial acantholysis resembling pemphigus foliaceus.
Actinic Prurigo
Actinic prurigo (hereditary polymorphous light eruption, Hutchinson summer prurigo) shares many features with polymorphous light eruption (PMLE). Some classifications include actinic prurigo as a severe variant of PMLE, although consistent clinical and histologic differences have been documented that permit separation of actinic prurigo as a distinct disorder. Actinic prurigo usually arises in childhood and is more common in women. Strong HLA-associated susceptibility has been documented. Most cases in the United States have been reported in native North Americans, but actinic prurigo also affects Mestizos (mixed Native Indian and European) in Central and South America and has also been reported in the United Kingdom. As a clinically polymorphous eruption, actinic prurigo exhibits variable histopathologic features. Characteristic histopathologic features include psoriasiform epidermal hyperplasia, papillary dermal edema, and perivascular dermatitis (often dense, lichenoid, and/or deep) with eosinophils. More variable findings include solar elastosis, orthokeratosis or parakeratosis, spongiosis, vacuolar interface changes, and plasma cells. Reactive lymphoid follicles are a variable but distinctive finding. Thus, although it is expected that some cases of actinic prurigo will be clinically and histologically indistinguishable from PMLE, the presence of psoriasiform hyperplasia, eosinophils, and lymphoid follicles favors actinic prurigo ( Fig. 3 ). Specific clinical features of actinic prurigo include conjunctivitis and cheilitis, and the typical ethnicity/ancestry.
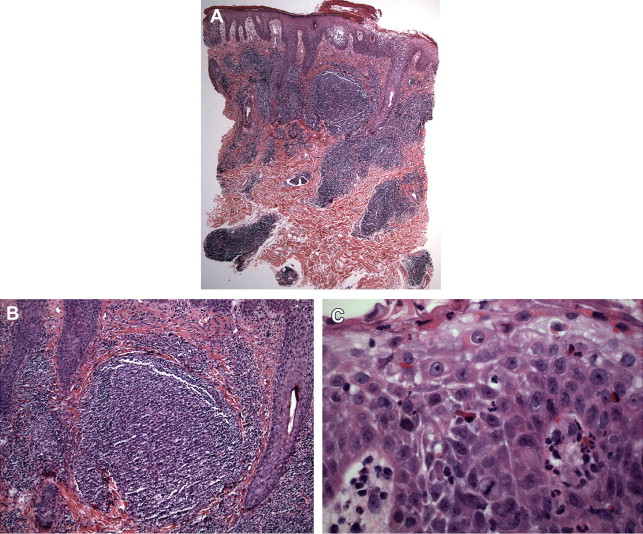
Interface dermatitis
Interface dermatitis may be subclassified based on the density of the associated inflammatory infiltrate—vacuolar (sparse) versus lichenoid (dense, band-like)—or based on the pattern of associated epidermal changes. LeBoit proposed the following five epidermal patterns: erythema multiforme–like (necrotic keratinocytes present in clusters and above the basal layer), lichen planus–like (basilar squamatization), irregular epidermal hyperplasia (eg, hypertrophic lichen planus), psoriasiform hyperplasia (eg, lichen striatus, lichen aureus), and atrophic (eg, poikilodermatomyositis).
A recently recognized pitfall in the interpretation of lichenoid dermatitis is the rare presence of small junctional nests of Melan-A immunopositive cells, thus presumed to be melanocytes. Clinical correlation is required to avoid an incorrect diagnosis of melanoma in situ in cases with these pseudonests. Final clinical-pathologic diagnoses have included lupus erythematosus, lichen planus, lichenoid phototoxic dermatitis, and lichenoid keratosis.
Cutaneous Lupus Erythematosus
Lupus erythematosus (LE) may exhibit sparse vacuolar change (usually acute or subacute cutaneous LE) or lichenoid change with denser deep perivascular and periadnexal extension of lymphocytes, including plasma cells, and follicular plugging (discoid variant of chronic cutaneous LE). Although perieccrine lymphocytes are distinctive, perineural involvement has also been documented. Marked papillary edema resembling polymorphous light eruption may rarely occur. Eosinophils are generally rare or absent in acute, subacute, and chronic cutaneous LE (with the exception of lupus profundus), and in dermatomyositis, graft-versus-host disease (GVHD), and pityriasis lichenoides.
Nonbullous neutrophilic variants of LE have been documented that exhibit dermal perivascular and interstitial neutrophils and leukocytoclastic nuclear debris (nuclear “dust”), without vasculitis or blister formation. Clinically, these lesions are more likely to present as pruritic urticarial papules and plaques compared with conventional cases of LE.
One potential diagnostic pitfall is the recognition of LE-like interface dermatitis induced by topical imiquimod therapy. Curiously, imiquimod also has been rarely reported to induce LE and its remission.
Paraneoplastic Dermatitis
Paraneoplastic dermatitis is not a defined clinical-pathologic entity but, rather, is a phrase that has been used by some clinicians to describe rare paraneoplastic reactions that are not readily classified. To date, the documented spectrum of paraneoplastic reactions includes paraneoplastic pemphigus (see next section) but also disorders that lack vesiculobullous lesions or mucositis clinically and acantholysis histologically. For example, unexplained spongiotic/eczematous dermatitis and “dermal hypersensitivity” reactions have been associated with systemic lymphoma.
Several recent reports have highlighted the presence of interface dermatitis in patients with thymoma, not only in humans but also in rabbits, cats, and dogs. A recent human case characterized as thymoma-associated multiorgan autoimmunity (TAMA) exemplifies the clinical and histologic resemblance to GVHD. In TAMA, a history of thymoma is definitional, and GVHD and drug reactions must be excluded by the clinical history. Gastrointestinal involvement has been a feature in all reported cases to date, with variable hepatic and cutaneous involvement. The skin lesions have been characterized as maculopapular (morbilliform), confluent nummular, or erythema multiforme–like. Oral involvement suggestive of paraneoplastic pemphigus may occur but, in contrast to paraneoplastic pemphigus, is not a hallmark of TAMA. Histologically, involvement of adnexal epithelium through an interface reaction with prominent necrotic/apoptotic keratinocytes represents an overlapping feature with GVHD, TAMA, and the lichenoid variant of paraneoplastic pemphigus (paraneoplastic autoimmune multiorgan syndrome) ( Fig. 4 ). Single-cell apoptosis is a common feature in TAMA-associated colitis and GVHD-associated colitis. A loss of T-regulatory function, as reflected by the loss of expression of transcription factor FOXP3 and autoimmune regular (AIRE) within intratumoral thymic T cells, might be indicative of a loss of tolerance to self-antigens in the pathogenesis of TAMA.
Histiocytic Necrotizing Lymphadenitis
Most individuals who develop histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto disease, Kikuchi disease) are young women who present with fever, pancytopenia, and lymphadenopathy, and in whom no skin lesions arise. However, the clinical and histologic spectrum of cutaneous lesions can be dramatic and have been better characterized over the past several years. The clinical course is generally benign and self-limited, but clinical correlation is required to exclude lupus erythematosus. Cases initially diagnosed as histiocytic necrotizing lymphadenitis have rarely evolved to systemic LE. Skin lesions may be maculopapular/morbilliform, nodular, or vesiculobullous. The mechanism of blister formation may be secondary to interface dermatitis or marked papillary dermal edema, and is accompanied by the distinctive presence of leukocytoclastic nuclear debris (leukocyte nuclear dust) without vasculitis or neutrophils ( Fig. 5 ). In both lymph node and skin, CD68-positive mononuclear cells (resembling plasmacytoid lymphocytes on H&E) were originally classified as plasmacytoid T cells or plasmacytoid monocytes. More recently, these lesional cells in the lymph node have been identified immunophenotypically as two populations of immature dendritic cells: plasmacytoid dendritic cells and myeloid dendritic cells.
Vasculitis and vasculopathy
Levamisole-Induced Vasculopathy
Levamisole-induced vasculopathy with ecchymosis and necrosis (LIVEN) has received a great deal of attention in the past couple of years after reports from multiple centers in North America attributing LIVEN to recreational use of cocaine intentionally contaminated with the veterinary antihelminthic agent, levamisole. The clinical presentation consists of necrosis and retiform purpura most commonly affecting the ears, cheeks, nose, digits, trunk, and proximal extremities, with variable progression to ulceration and eventual remission. Patients have a history of recreational cocaine use (sniffed, snorted, or smoked) and present with associated neutropenia, positive antineutrophil cytoplasmic antibody (ANCA) serologies (both p-ANCA and c-ANCA), and hypocomplementemia. Antibodies against neutrophil granules (neutrophil elastase, myeloperoxidase, lactoferrin, cathepsin G, proteinase 3) are also present. In particular, the presence of human neutrophil elastase antibodies has been proposed to be a sensitive and specific feature of LIVEN that distinguishes it from autoimmune vasculitis. Agranulocytosis with associated myeloid hypoplasia in the bone marrow may be associated with potentially fatal secondary systemic bacterial or fungal infection. Histologically, small vessel thrombosis with or without leukocytoclastic vasculitis is characteristic ( Fig. 6 ). In patients with vasculitis, direct immunofluorescence demonstrates vascular immune complex deposition. Extensive perivascular and interstitial neovascularization may be seen. These clinical and histologic features are not specific for LIVEN, and therefore additional clinical and laboratory evaluation may be required to definitively assess for other causes of coagulopathy (eg, antiphospholipid antibody syndrome, cryoglobulinemia, warfarin-induced skin necrosis) or other causes of vasculitis. LIVEN manifesting as purpura on the earlobes and cheeks has been documented in children receiving levamisole as adjuvant therapy for nephrotic syndrome.
Macular Lymphocytic Arteritis and Lymphocytic Thrombophilic Arteritis
Macular lymphocytic arteritis (MLA) and lymphocytic thrombophilic arteritis (LTA) are designations that seem to reflect nearby points along the spectrum of a single rare but distinctive clinical-pathologic entity whose relationship, if any, to cutaneous polyarteritis or autoimmune connective tissue disorders remains uncertain. Most patients to date have been young or middle-aged adult women, with macular, livedoid, or minimally indurated nonulcerated lesions usually on the lower extremities bilaterally. The clinical course is chronic and benign, and the clinical evaluation, by definition, falls short of a connective tissue disorder or coagulopathy, such as antiphospholipid antibody syndrome, although low-titer anticardiolipin antibodies are frequently documented in these individuals. The histologic common denominator is a lymphocytic arteritis centered at the dermal-subcutaneous junction associated with a thick fibrin ring (resembling a strawberry donut) partly occluding the lumen of the arteriole (endarteritis obliterans). A sparse to moderately dense lymphocytic infiltrate with nuclear dust is present, but neutrophils and eosinophils are minimal or absent. Elastin stains usually demonstrate an intact elastic lamina.
Vesiculobullous disorders
Grover Disease (Transient Acantholytic Dermatosis)
Histologic variants of Grover disease may resemble Darier disease (acantholytic dyskeratosis), pemphigus vulgaris (suprabasilar acantholysis), pemphigus foliaceus (intragranular acantholysis), Hailey-Hailey disease (full-thickness acantholysis), or spongiotic dermatitis. Grover disease may rarely coexist with herpes simplex virus (HSV); the viral cytopathic changes may be obscured, particularly by acantholytic dyskeratosis ( Fig. 7 ). The demonstration of HSV-I immunohistochemical positivity in a case of Grover disease, and in rare cases of Darier disease, pemphigus vulgaris, and bullous pemphigoid, suggests the possibility of occult colonization by herpesvirus in some examples of these diseases. Clinical superinfection analogous to eczema herpeticum may also occur in Grover disease.
Papular Genitocrural Acantholysis
Papular genitocrural acantholysis (papular acantholytic dyskeratosis of the vulva) is included in the 2006 International Society for the Study of Vulvar Dermatoses (ISSVD) Classification for Vulvar Dermatoses for papular lesions confined to the genitocrural region that show the epidermal reaction pattern of acantholytic dyskeratosis. Immunofluorescence studies are negative. This benign disorder may occur in adults or children, and is believed to represent cases reported in the past as Darier disease (keratosis follicularis) localized to the vulva.
Paraneoplastic Pemphigus
Paraneoplastic pemphigus was the subject of a recent review in Dermatologic Clinics , but selected aspects are also reviewed here. Since its original description by Anhalt and coworkers in 1990, the spectrum of paraneoplastic pemphigus subsequently expanded to include lichenoid variants. The additional recognition of systemic involvement (often pulmonary) resulted in a broader classification under the heading of paraneoplastic autoimmune multiorgan syndrome proposed by Grando and coworkers. This broadened disease spectrum encompassed under the heading of paraneoplastic pemphigus/paraneoplastic autoimmune multiorgan syndrome may explain the elusive nature of diagnosis. Classic cases of paraneoplastic pemphigus present with oral involvement, an associated internal malignancy, biopsies that show combined acantholytic and interface changes, perilesional direct immunofluorescence (DIF) showing concomitant intercellular (cell surface) and basement membrane zone (BMZ) deposition of IgG and C3, and confirmatory serum studies (indirect immunofluorescence, enzyme-linked immunosorbent assay [ELISA], immunoblotting, immunoprecipitation) for IgG antibodies directed against many or all of the recognized components of the antigen complex that contain the self-targets in paraneoplastic pemphigus: desmoplakin I (250-kd), bullous pemphigoid antigen 1 (230-kd), desmoplakin II-envoplakin (210-kd), periplakin (190-kd), and a 170-kd protein. The existence of several reports of nonclassical presentations of paraneoplastic pemphigus emphasizes the need for careful correlation between the clinical, H&E, DIF, and serum studies in the evaluation of patients with pemphigus. Nonclassical examples include cases with negative serum studies, no malignancy, either pure acantholytic or pure lichenoid histology, and negative or nonclassic DIF findings.
Granulomatous dermatitis
Granulomatous dermatitis is traditionally subclassified into sarcoidal, tuberculoid, palisaded, interstitial, and foreign body types. During medical school, the first author reported a novel finding of “granuloma initiation factor” using a Schistosoma mansoni –murine model ; in retrospect, this was a very focused and relatively narrow concept of a diverse spectrum of diseases with granulomatous inflammation. The past decade saw an ever-broadening spectrum of granulomatous reactions that sometimes seems intent on teaching more by breaking the established rules. At the same time, the increasing use of immunosuppressive therapy has resulted in a resurgence of opportunistic infections, of which fungi, mycobacteria, and protozoa stereotypically induce a suppurative neutrophilic granulomatous host response.
Sarcoid
Cutaneous sarcoid is clinically protean but characterized histologically by sarcoidal granulomas composed of nodules of epithelioid histiocytes and sparse lymphocytes (“naked granulomas”). However, Ball and coworkers showed a significantly broader spectrum of findings in clinically confirmed cases, including tuberculoid granulomas, interstitial granulomatous infiltrates, and associated lichenoid interface reactions. Perineural granulomas have been documented, as has evolution from palisaded neutrophilic and granulomatous dermatitis (see next section). Perigranulomatous fibroplasia has been reported to be a distinctive feature of subcutaneous sarcoid.
Immunodeficiency Syndrome–Associated Granulomatous Dermatitis
Granulomatous dermatitis may occur in a variety of immunodeficiency syndromes, including ataxia telangiectasia, chronic granulomatous disease, severe combined immunodeficiency, common variable immunodeficiency, X-linked hypogammaglobulinemia, and familial hemophagocytic lymphohistiocytosis ( Fig. 8 ). The pattern may be sarcoidal, tuberculoid, necrotizing, or palisaded.
Related posts:
 Cytogenetic and Mutational Analyses of Melanocytic Tumors
Cytogenetic and Mutational Analyses of Melanocytic Tumors
 Dermatology Clinics
Fibrous and Fibrohistiocytic Neoplasms
Current Understanding of Cutaneous Lymphoma
Direct Immunofluorescence Testing in the Diagnosis of Immunobullous Disease, Collagen Vascular Disease, and Vascular Injury Syndromes
New Directions in Dermatopathology
Dermatology Clinics
Fibrous and Fibrohistiocytic Neoplasms
Current Understanding of Cutaneous Lymphoma
Direct Immunofluorescence Testing in the Diagnosis of Immunobullous Disease, Collagen Vascular Disease, and Vascular Injury Syndromes
New Directions in Dermatopathology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


