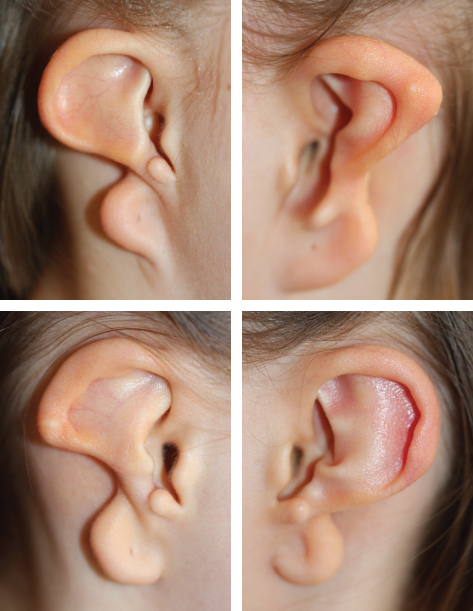
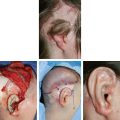
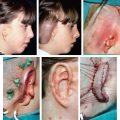
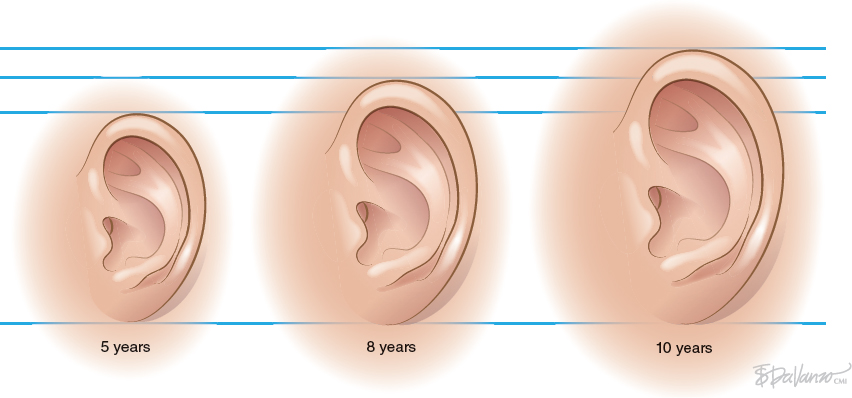
CHAPTER 4 Patients with a microtia often present for the first consultation in the neonatal period or as a young infant; thus surgeons must have a treatment plan ready to discuss with the parents. For autologous reconstructions, we choose to delay the reconstruction of the ear until patients are 9 or 10 years of age for several important reasons: 1. Sufficient cartilage is necessary to create all the contours of the ear, and the thoracic development is usually not adequate until after patients are 8 years of age. 2. It is impossible to guess the future size of an ear in unilateral microtia; thus we need to wait until the growth of the normal ear is almost complete. After routine measurements of hundreds of patients, we have found that this occurs by 9 or 10 years of age. 3. Finally, the reconstruction of an ear is a personal decision and should not be undertaken until the child is mature enough to express his or her desire to have a new ear. Fig. 4-1 A patient’s ear is shown at 5, 8, and 10 years of age. By 10 years of age, it has grown to adult size. Microtia literally means “small ear.” Clinically, there is a very wide spectrum of morphologic findings of the microtic ear, ranging from complete absence of the ear remnant (anotia) to mild anomalies in the shape of the ear. Fig. 4-2 This 16-year-old patient has a very nice, normal face. She presents with an isolated right microtia. Microtia: • Occurs more frequently in males • Occurs more commonly on the right side • Is more commonly unilateral than bilateral • Has an overall incidence of 3:10,000 • Has a higher incidence in Hispanics, Asians, American Indians, and Andeans Our goal is not to list all the possible associations of anomalies of the face. Rather, we will focus on the particular surgical problems frequently encountered that affect the surgeon’s ability to correctly reconstruct a microtic ear. Although microtia most commonly occurs as an isolated unilateral malformation, it can be associated with several syndromes. The most frequent association is with hemifacial microsomia (HFM) (also known as otomandibular syndrome) and Goldenhar syndrome. These conditions form part of a larger grouping of syndromes known as the occuloauriculovertebral spectrum. The causes for these slightly different syndromes are not known. However, the tissues derived from the first and second branchial arch are overwhelmingly affected and, as a result, the syndromes are inherently linked to microtia. To help understand and classify the most frequent facial abnormalities of HFM, the OMENS classification has been developed.
Congenital Malformations
DETERMINING THE CONTEXT OF MICROTIA RECONSTRUCTION
ISOLATED MICROTIA

HEMIFACIAL MICROSOMIA
O: | Orbital asymmetry |
M: | Mandibular hypoplasia |
E: | Ear deformity |
N: | Nerve involvement |
S: | Soft tissue involvement |
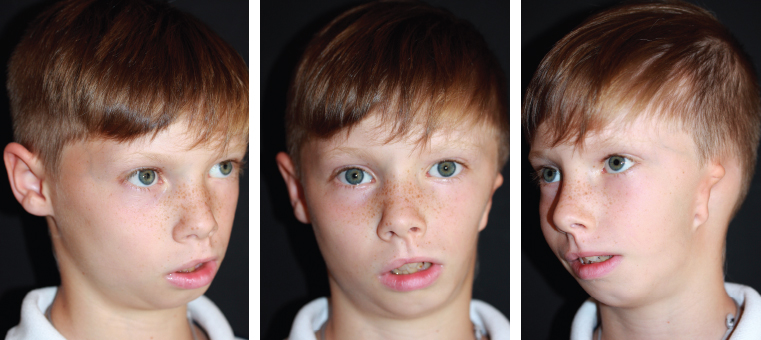
Fig. 4-3
This 9-year-old patient has HFM (see video 4-1).
In our series we have identified two pathognomonic signs that indicate a diagnosis of HFM, even when the face seems normal:
1. Absence of the preauricular sideburn
2. Presence of a chondroepidermal pouch

Absence of the Preauricular Sideburn
Although facial asymmetry may not be immediately obvious from the frontal view, the hallmark features of HFM are present when analyzing the auricular region. In these cases the preauricular sideburn is absent.
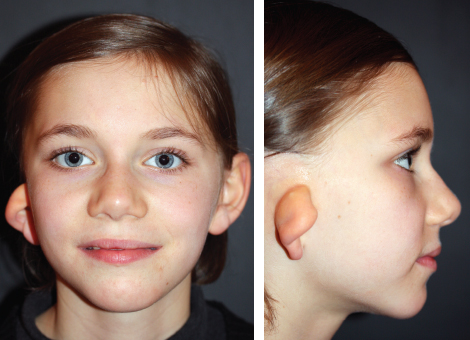
Fig. 4-4
These two patients seem to have a symmetrical face from the frontal view, but the characteristic absence of a sideburn helps to make a diagnosis of HFM.
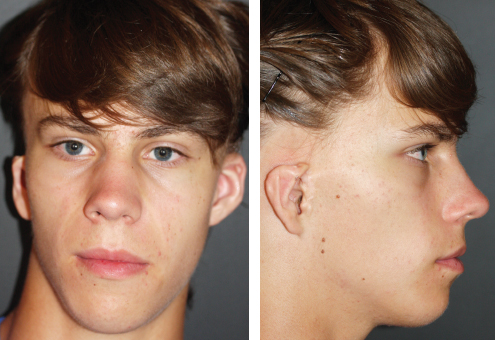
Fig. 4-5
These two patients seem to have a symmetrical face from the frontal view, but the characteristic absence of a sideburn helps to make a diagnosis of HFM.
Presence of a Chondroepidermal Pouch
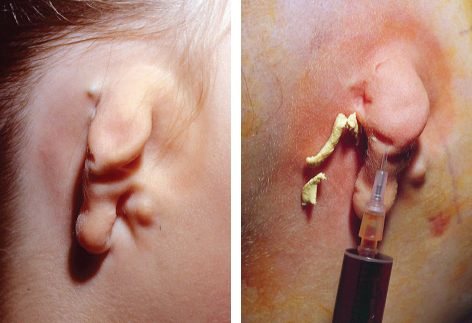
Fig. 4-6
A chondroepidermal pouch is another pathognomonic feature of HFM. Clinically, this pouch is a large cutaneous invagination within the microtic remnant, which has the peculiarity of being surrounded by fibrocartilage.
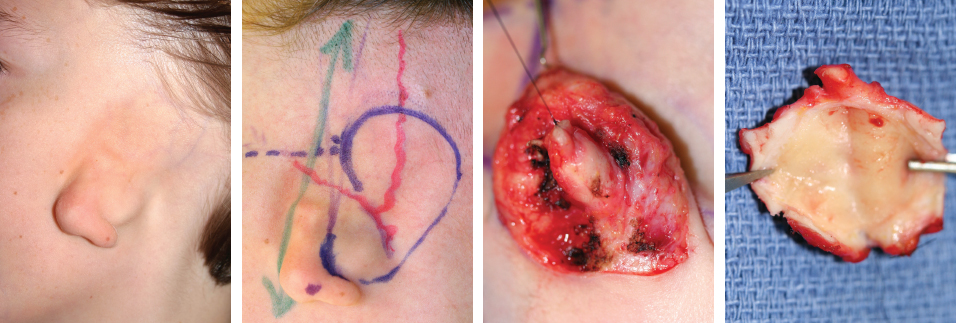
Fig. 4-7
This is a typical case of HFM with a chondroepidermal pouch and the absence of a sideburn. The opening of a pocket was seen in the undersurface of the remnant, and this was excised en bloc without trying to wash out the contents. The opening was sutured closed before starting the surgical preparation to prevent contamination of the field.
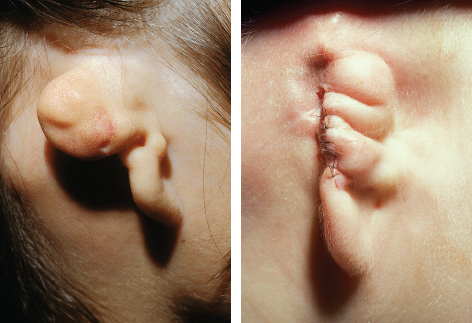
Fig. 4-8
Any history of previous infection or signs of active inflammation or malodorous contents necessitate formal excision of the pouch before reconstruction of the ear.
The superficial temporal artery is often ectopic in HFM (see Chapter 2, Fig. 2-19). However, in contrast to Treacher Collins–Franceschetti (TCF) syndrome, HFM does not have a clear genetic basis, and the etiologic factors remain unknown. The vascular insult theory may explain the unilaterality and asymmetry of the disorder, which is bilateral in less than 20% of cases. Arrest of neural crest cell migration is also a proposed mechanism.
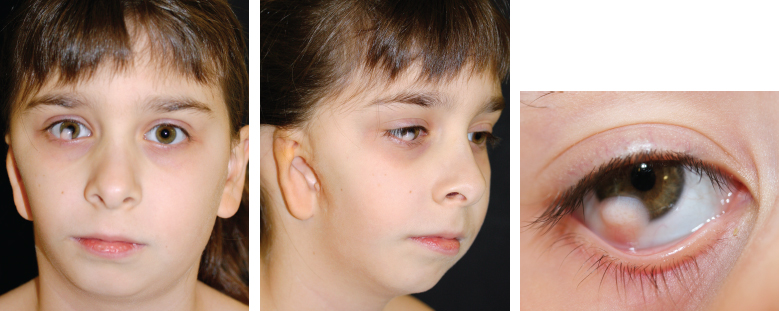
Goldenhar syndrome may include abnormalities of the cervical vertebrae and the eye and facial hypoplasia. The presence of an epibulbar dermoid is pathognomonic. Determining the ideal position of the reconstructed ear is challenging in patients with HFM, because their face is asymmetrical, and the normal landmarks are not available on the affected side. This will be discussed in detail later in this chapter. Routine assessment in a multidisciplinary clinic will help to determine the ideal timing of the mandibular correction.
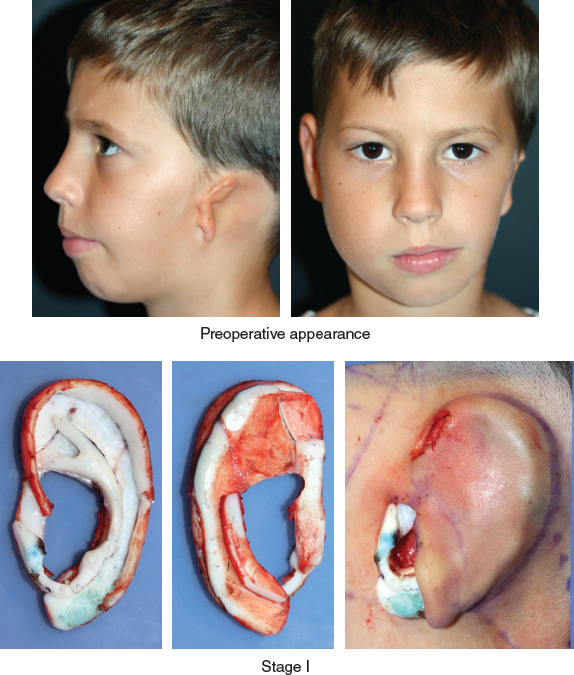
Fig. 4-10
This patient with HMF was seen in a multidisciplinary consultation. A decision was made to delay definitive mandibular reconstruction until after skeletal maturity and to use serial fat injections to correct the soft tissue deficiency of the cheek.
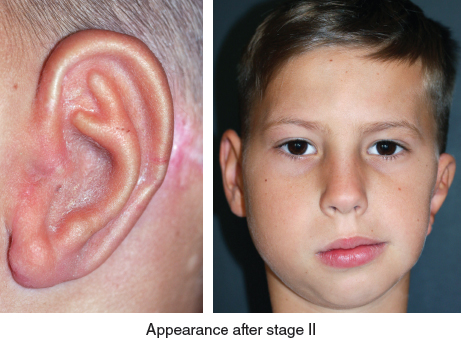
After a two-stage auricular reconstruction (type 3b skin approach, TYPE I PI, PII projection, and type D second stage), the ear still seems a little lower than the other one.
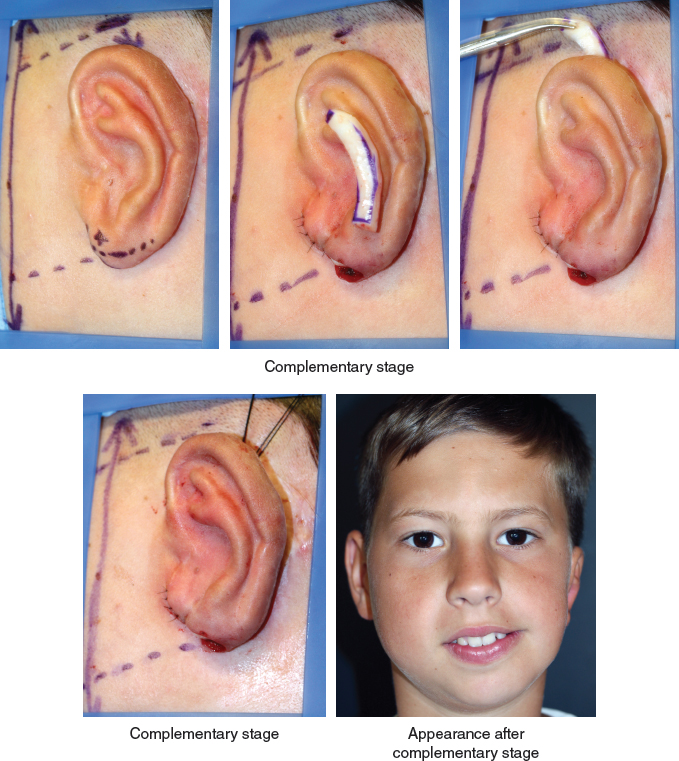
A complementary stage will correct the position of the ear, not only reducing the lobule but also elevating the ear. The projection of the ear was improved by inserting an additional cartilage graft in a tunnel behind the antihelix, and the lower position was corrected by freeing the entire posterior surface of the framework and fixing it with a strong anchoring stitch as shown. This case demonstrates that correctly positioning the ear in the vertical plane is essential, particularly in cases of facial asymmetry.
TREACHER COLLINS–FRANCESCHETTI SYNDROME
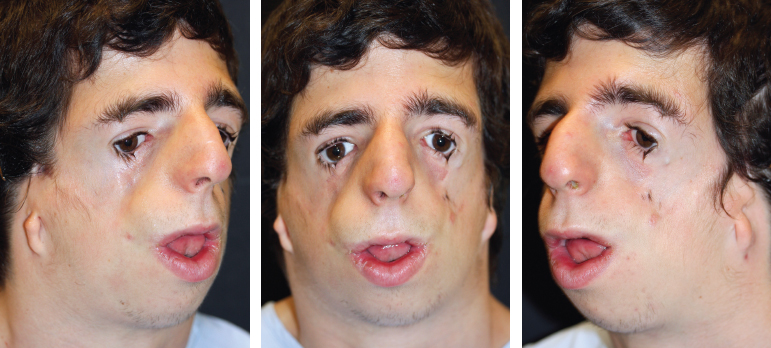
Fig. 4-11
TCF syndrome is an inherited autosomal dominant condition frequently associated with bilateral microtia and a characteristic facial deformity (see video 4-2). An almost universal finding is notching and hypoplasia of the lower lid with downslanting palpebral fissures. The middle ear is commonly atretic, resulting in deafness from bilateral conductive hearing loss. The nasopharynx is affected by palatal clefting and choanal atresia. The lateral view shows a characteristic birdlike appearance caused by a prominent, beaked nose and a retruded mandible.

Dr. Paul Tessier described the underlying bony deformity of TCF syndrome as a cleft of one or all of the bony fusion points of the zygoma, and named them laterofacial clefts 6, 7, and 8.* The presence of all three clefts results in agenesis of the zygomatic bone altogether and unrestricted maxillary growth, yielding the classic beaked-nose appearance.
TCF syndrome is almost always symmetrical. A review of our series in 2014 showed that of 82 patients, 85% had bilateral microtia, and 75% of these had the bilateral lobular type.
Pathognomonic Features of Treacher Collins–Franceschetti Syndrome
Our study identified three pathognomonic features of TCF syndrome that do not tend to appear in the other laterofacial clefting syndromes, such as HFM and Goldenhar syndrome.
1. An anteriorly projecting preauricular sideburn (62%)
2. Ectopic course of the superficial temporal artery (75%)
3. Low hairline (25%)
Anteriorly Projecting Preauricular Sideburn
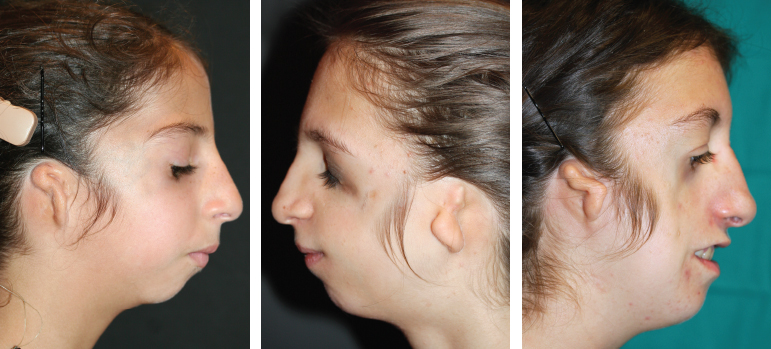
Fig. 4-12
An anteriorly projecting preauricular sideburn is a very common finding. It is often associated with a low hairline. Together these clinical findings only presented in cases with severe facial deformity; thus we consider them a marker of severity.
Ectopic Course of the Superficial Temporal Artery
An ectopic course of the superficial temporal artery was a very consistent feature of TCF syndrome (75%), occurring equally in severe and mild forms.
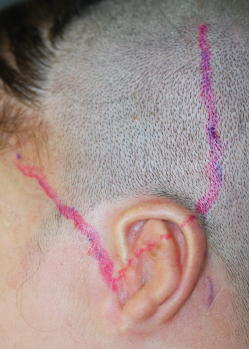
Fig. 4-13
We routinely perform Doppler ultrasonography during the first surgery to determine the course of the superficial temporal artery. In TCF syndrome this is crucial, because the artery is often ectopic and must be preserved while the surgeon undertakes superficial dissection of the skin pocket to insert the framework.
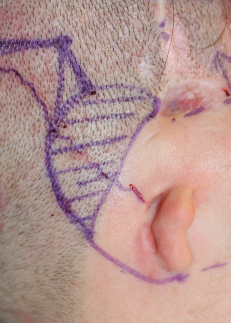
This patient had a coronal approach before ear reconstruction, but the ectopic course of the artery was serendipitous, because it preserved blood supply to the temporoparietal fascia. Doppler ultrasonography should be performed and this axial vessel protected for any surgery in the temporal area in patients with microtia.
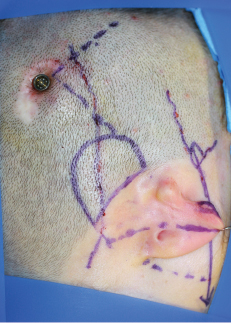
Fig. 4-15
Doppler examination shows the ectopic course of the artery, which traverses the zone of the bone-anchored hearing aid (Baha, Cochlear). We advise caution in elevating the temporal fascia in a scarred environment (see Chapter 5).
Low Hairline
In patients with a low hairline, a temporofascial flap is indicated during the first stage to cover the upper part of the ear.
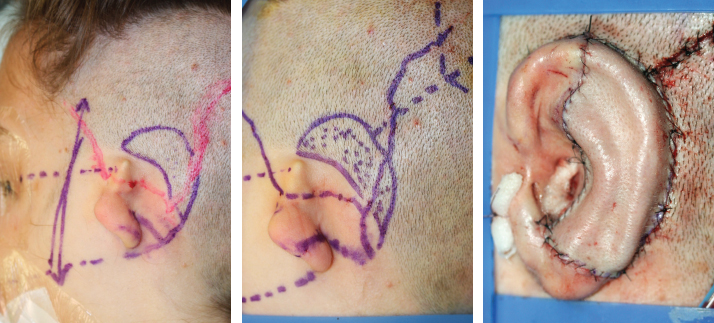
Fig. 4-16
After the scalp is shaved, Doppler ultrasonography is routinely performed. The position of the ear should not be compromised. In this case a Y-shaped incision with short fascia was used because of the location of the ear.
Specific Surgical Problems
We will discuss a few key surgical problems that are particularly relevant to TCF syndrome.
Management of the Airway in Treacher Collins–Franceschetti Syndrome
Patients with TCF syndrome should be treated as having difficult airways until proved otherwise. We routinely perform fibroscopic intubation of all TCF patients because of the high frequency of choanal atresia and mandibular retrognathism. Obstructive sleep apnea, which is a life-threatening concern, should be evaluated by specialist services.
Multidisciplinary Approach
Multiple operations require a multidisciplinary, staged approach.
Patients regularly request ear reconstruction before treatment for the rest of their facial deformities. To these patients the ears are missing, whereas their other facial features are present but abnormal. Thus patients commonly have bicoronal approaches to address the midfacial cleft before 9 or 10 years of age. As previously shown, the coronal approach can damage the temporoparietal fascial flap and superficial temporal artery; thus we strongly urge craniofacial surgeons to keep this in mind and try to preserve the fascia if at all possible. Furthermore, our multidisciplinary team has found that serial episodes of fat grafting of the malar and lower eyelid in young TCF patients may prevent the need for osseous midfacial reconstruction altogether, preventing the need for a coronal approach.
Bone-anchored Hearing Aid Placement
Because of the high frequency of bilateral auditory canal and middle ear atresia, bone-anchored hearing aids are routinely placed in patients with TCF syndrome. The position of this device can severely impede the positioning and reconstruction of the ear. Surgeons should carefully place the device as posteriorly and inferiorly as possible, as shown here.
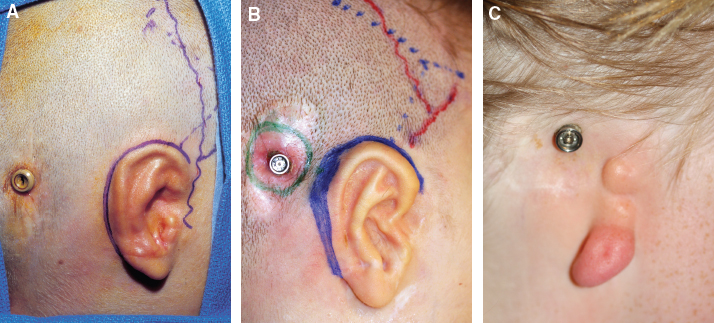
Fig. 4-17
This bone-anchored hearing aid is perfectly placed and did not affect the first or second stage (A). The second device shown is positioned too anteriorly and has a large area skin grafted around the abutment, which is an antiquated technique (B). New implants do not require skin grafting. The third device shown is erroneously placed, and the abutment must be relocated before the ear can be reconstructed (C).
Low Position of the Ears
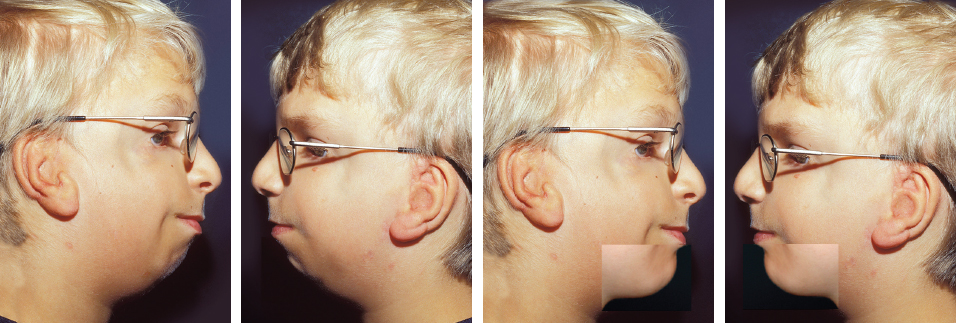
Fig. 4-18
In patients with TCF syndrome, the ear may appear to be in a low position after surgery. This is an apparent finding because of the retruded position of the mandible. When compared with other facial features, the ear is in the correct position. This is seen by predicting the future position of the mandible.
COMPLEX FACIAL MALFORMATIONS
Microtia may present as part of a more severe and complex craniofacial malformation. A multidisciplinary team must take charge of the care of the patient. Each of the competing priorities of the patient is combined into a reasonable chronologic surgical plan. Surgeons must consider the desires of the patient.
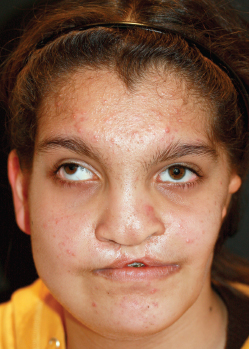
Fig. 4-19
This patient presented with a repaired bilateral cleft lip associated with a severe hypoplasia of the left side of the face that was corrected elsewhere with the insertion of a dermal fat graft from the inguinal area.
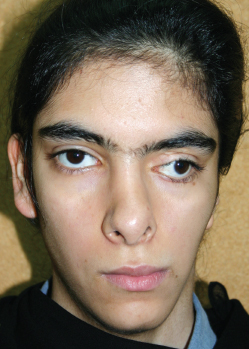
Fig. 4-20
This patient had a severe orbital asymmetry that could benefit from a craniofacial solution, but the patient was only motivated to have ear reconstruction.
GENETIC CONSIDERATIONS
Most isolated microtia cases are sporadic with a multifactorial or polygenic cause. In syndromic and familial cases of microtia, a Mendelian inheritance pattern is more likely, and several genes have now been identified in syndromes associated with microtia.
Isolated Microtia
Although the genetic basis for isolated microtia has not been precisely determined, the following evidence has been reported:
1. Concordance was higher in identical twins than in nonidentical twins.
2. Several syndromes are associated with microtia, for which the genetic basis has been identified, as previously mentioned.
3. Many familial cases are reported the literature and also in our series.