Collagens, Elastic Fibers, and Other Extracellular Matrix Proteins of the Dermis: Introduction
|
Introduction
The extracellular matrix (ECM) is a complex network of different components, responsible for determining and maintaining tissue architecture, and for mediating a number of important biological events. It is composed of a large number of diverse protein families, each constituted by many different individual members. These include the collagens, encoded by 42 different genes, elastin and associated microfibrillar proteins, fibronectin, proteoglycans, and many more molecules. (See Table 63-1.)
Proteins | Number of Genes | Number and Name of Proteins |
|---|---|---|
Collagens | 42 | 28; collagens type I to XXVIII |
Elastin | 1 | Several splice variants |
Fibronectin | 1; 20 splice variants | |
Fibulins | 7 | 7; Fibulin 1–7 |
Fibrillins | 3 | 3; Fibrillin 1–3 |
Laminins | 11 | 15; LM-111, LM-332, LM-511, etc. |
Matrilins | 4 | 4; Matrilins 1–4 |
Nidogens | 2 | 2; Nidogen 1 and 2 |
Tenascins | 4 | 4; Tenascin-C, -X, -R, and -W |
Thrombospondins | 5 | 5; Thrombospondin 1–5 |
Vitronectin | 1 | 1 |
Although all of these proteins are genetically, structurally, and biologically diverse, a common denominator is that most of them have a modular structure, and they are composed of one, a few, or several copies of a limited set of individual structural modules, also called domains.1 These can be combined in multiple ways giving rise to proteins as diverse as fibrillin and laminin (Fig. 63-1). Specific functions have been unraveled for some of these domains, for instance, interaction with other ECM proteins, cell adhesion-promoting activity, cytokine trapping, and regulation. Therefore, the ECM has a critical role for many cellular functions, including proliferation, survival, polarity, differentiation, expression of specific genes, and migration.2–4 All different cell types, such as mesenchymal, epithelial, and endothelial cells, and also inflammatory and tumor cells, participate in the production of distinct ECM macromolecules, and are all influenced by interactions with these compounds. It is well established that the ECM determines the biophysical properties of connective tissues. More recently it became clear that, conversely, stiffness and compliance of connective tissues are important factors for the regulation of cellular functions.5–8
Figure 63-1
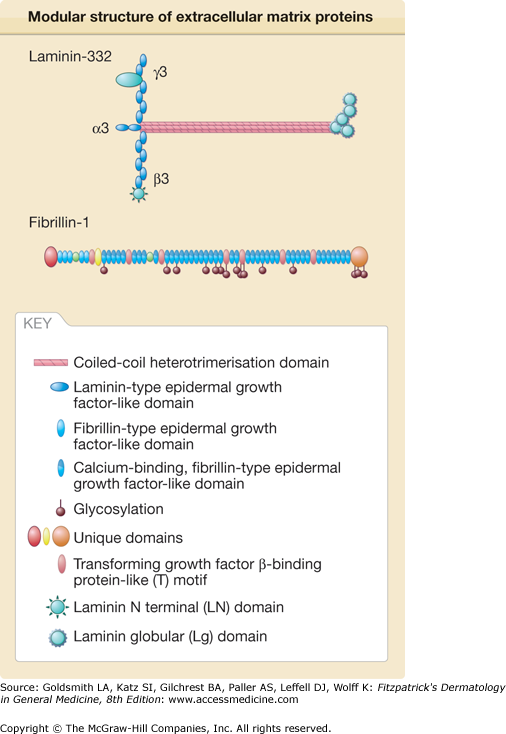
Modular structure of extracellular matrix (ECM) proteins. ECM proteins are composed by a few individual structural modules, such as fibrillin- and laminin- type epidermal-growth factor-like modules, von Willebrand factor A domain, follistatin-like and EF-hand Ca2+-binding motif or coiled-coil oligomerization domains. These modules can be combined in multiple ways giving rise to proteins as diverse as fibrillin or laminin displayed here as examples. In addition, ECM proteins can be formed of a single polypeptide chain, as fibrillin, or of several polypeptide chains. For instance, laminin 332 is formed by three polypeptide chains, the a3, b3 and g2 chains.
Collagens
The proteins of the collagens family are the main structural components of the connective tissues and the major extracellular proteins of the human body. In human skin, collagens comprises approximately 80% of the dry weight of the dermis. The classical and first recognized physiologic role of collagens in the skin is to provide tensile properties that allow the skin to serve as a protective organ against external trauma. In addition, it is clear that collagens displays important biological properties regulating multiple cellular and tissue activities.
The bulk of collagens in dermis is deposited as large bundles of regularly oriented fibers composed of fibrils that are aligned in a parallel manner, which results in a pattern of cross-striations that can be visualized by electron microscopy (Fig. 63-2). The most prominent cross-striations appear as repeating bands spaced approximately 70 nm apart. The major component of these fibrils is type I collagens, the first identified member of the collagens family and the most abundant collagens in the dermis and most other connective tissues.9–11 The characteristic structural features of collagens were deduced from the study of type I collagens, which is therefore considered as the prototype of the collagens family.

The type I collagens molecule has an approximate molecular mass of 290 kDa and is composed of three polypeptide chains, each approximately 94 kDa. These three polypeptides, known as the α chains, are coiled around each other much like strands of rope, so that the collagens monomer has a triple-helical structure. This conformation gives the molecule a rigid, rod-like shape with approximate dimensions of 1.5 × 300 nm (see Fig. 63-3). The special structure of the collagens triple helix is largely explainable by the unusual amino acid composition of the α chains. Specifically, each α chain of type I collagens has approximately 1,000 amino acids, and glycine (Gly), the smallest amino acid, accounts for approximately one-third of the total number of amino acids, evenly distributed along the polypeptide. Consequently, the polypeptide chains of type I collagens can be described as repeating triplets represented as (Gly-X-Y)333. Although the X and Y positions of the triplets can be occupied by any amino acid, they are often occupied by proline and hydroxyproline, respectively. These two amino acids account for approximately 22% of the total amino acid composition of type I collagens. The relatively high content of these amino acids and the characteristic distribution of glycine in every third position are necessary for the triple-helical conformation of the collagens molecule, and hydroxyproline plays a critical role in stabilizing the triple helix at body temperature. The triple-helical conformation defines the so-called collagenous domain, and it gives type I collagens many of its unique properties.12 In particular, it is essential for assembly into fibrils. Mutations that affect the formation of the stable triple helix prevent collagens from forming fibers, which results in serious defects of connective tissue function and a spectrum of clinical phenotypes.13–15
Figure 63-3
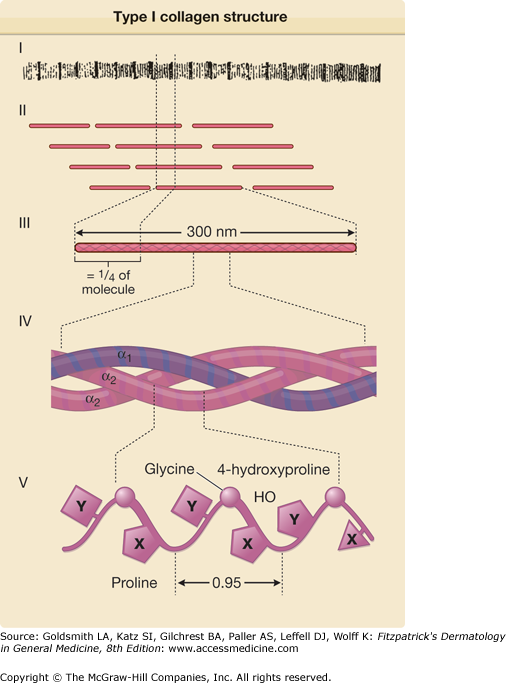
Schematic representation of the structure of type I collagens. The collagens fibers (I), which on electron microscopy demonstrate a repeating periodicity (see Fig. 63-2), are composed of individual collagens molecules aligned in a quarter-stagger arrangement (II). Each type I collagens molecule is an approximately 300-nm-long rod-like structure (III) consisting of three individual polypeptides, known as α chains, which are twisted around each other in a right-handed triple helix (IV). Each chain is composed of amino acids in a repeating Gly-X-Y sequence (V); as indicated, the X position is frequently occupied by a prolyl residue and the Y position is frequently occupied by a 4-hydroxyprolyl residue. The individual α chains have a left-handed helical secondary structure with a pitch of 0.95 nm. (From Uitto J et al: Collagens structure, function, and pathology. In: Progress in Diseases of the Skin, vol. 1, edited by R Fleischmajer. New York, Grune & Stratton, 1981, p. 103, with permission.)
Collagens comprises a family of closely related yet genetically distinct proteins. In the human genome, there are as many as 42 different genes encoding α chains with variable amino acid sequences. These α chains correspond to at least 28 different types of collagens, which have been assigned Roman numerals I to XXVIII (Table 63-2). These are subdivided into several subfamilies, mainly according to the length and number of collagenous and noncollagenous domains. In addition to well-characterized collagens, short triple-helical collagenous segments are present in other proteins, including acetyl cholinesterase, the C1q component of the complement system, gliomedin, pulmonary surfactant proteins, macrophage scavenger receptors, emilins (elastin microfibril interface-located glycoproteins), and ectodysplasin A, a product of the gene mutated in X-linked anhidrotic ectodermal dysplasia.16 However, these proteins are not included in the collagens family because the collagenous domains are not a predominant part of the molecules, and the proteins do not function primarily as structural components of the ECM of connective tissue.
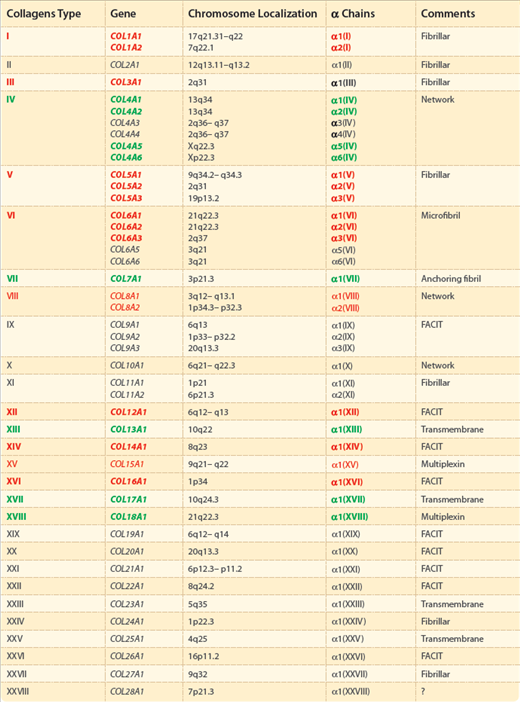
The genetically distinct collagens are distributed into several classes, mainly according to the length and number of triple-helical collagenous segments and noncollagenous domains present in the molecule, and on the basis of the architecture of their assembly in tissues (see Table 63-2). A classification and an overview of the major collagens types contributing to skin physiology and pathology are presented in the following paragraphs. Some collagens types are not discussed in detail because they do not appear to be present in the skin in significant quantities, or they do not seem to contribute to the physiologic properties of dermis, or because information is still lacking.
The first class groups the fibril-forming collagens types I, II, III, V, XI, XXIV, and XXVII. Except for collagens II, XXIV, and XXVII that were not detected in skin, all other fibril-forming collagens types are expressed in the dermis. These collagens types consist of a single 300-nm long collagenous domain, with very short nontriple-helical extensions (Fig. 63-4), and they assemble into relatively large fibrils.11,12,17 Type I collagens, the most widely distributed and the most extensively characterized form of collagens, accounts for approximately 80% of the total collagens of adult human dermis. It is formed by two identical α chains, designated α1(I), and a third chain, called α2(I), that is clearly different in its amino acid composition. Thus, the chain composition of type I collagens is [α1(I)]2α2(I). Type III collagens represents approximately 10% of the total collagens in the adult human dermis.18 It is composed of three identical α1(III) chains, distinguished from the chains of type I collagens by a relatively high content of hydroxyproline and glycine and the presence of one cysteine residue. Collagens types I and III form the relatively broad extracellular fibers that are primarily responsible for the tensile strength of the human dermis. Mutations in the type I and III collagens genes can result in connective tissue abnormalities in the skin and joints, among other tissues, in different forms of Ehlers–Danlos syndrome and fragility of bones in osteogenesis imperfecta.13–15 Type V collagens forms a subfamily of similar interrelated collagens resulting from various assemblies of three different types of α chains, α1(V), α2(V), and α3(V). The predominant form in the skin is [α1(V)]2α2(V), where it represents less than 5% of the total collagens. In dermis, type V collagens is associated with major collagens fibers consisting of type I and III collagens, and type V collagens has been postulated to regulate the fiber diameter during fibrillogenesis. The importance of type V collagens has been demonstrated by the discovery of mutations in type V collagens genes in patients with classical autosomal dominant forms (types I and II) of Ehlers–Danlos syndrome.19 It is of interest that a clinically similar, classical type of Ehlers–Danlos syndrome can be based on the absence of tenascin-X expression, which leads to an autosomal recessive form of the disease.20 Tenascin-X is developmentally associated with collagens fibrillogenesis, and its absence could explain the presence of defective collagens fibers similar to those found in patients with mutations in type V collagens genes.
Figure 63-4
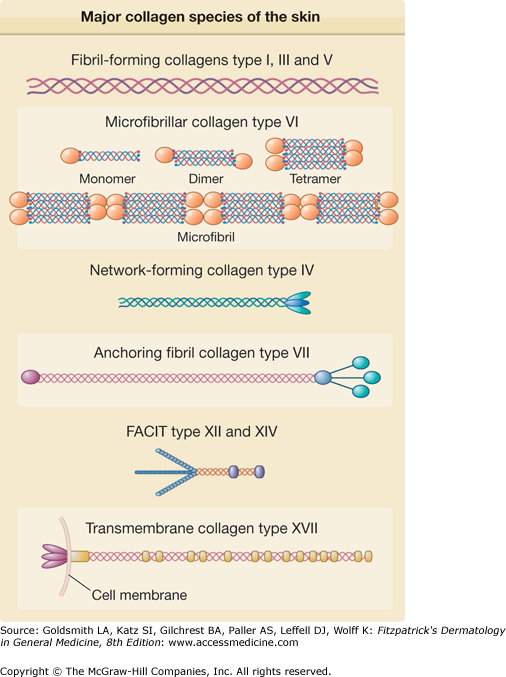
The major collagens species of the skin. According to several features, including length and number of collagenous and noncollagenous domains and supramolecular assemblies, the collagens molecules are grouped in different families. Type I, III, and V collagens have the characteristic rod-like structure and form the large collagens fibrils of the dermis. Type VI collagens molecules are arranged following a unique microfibrillar pattern of thin and long aggregates in the dermis. Also present in the dermal extracellular matrix are two FACITs (fibril-associated collagens with interrupted triple helices), type XII and XIV collagens associated to the large collagens fibrils formed by type I, III, and V collagens. Type IV collagens is ubiquitous of basement membranes where it forms large hexagonal networks. Two further collagens, type VII and type XVII, are specific of the dermal–epidermal junction, where type VII collagens is a component of the anchoring fibrils, while the ectodomain of transmembrane type XVII collagens is located in the anchoring filaments anchoring basal keratinocytes of the epidermis to the underlying basement membrane.
Another class comprises the network-forming collagens types IV, VIII, and X, which are, however, very different structurally (Fig. 63-4). Type IV collagens is a typical component of basement membranes, where it forms a lattice rather than the fibers characteristic of dermal collagens. Six different α (IV) chains exist, α1(IV) to α6(IV), and participate in the assembly of heterotrimers. In human skin, α1(IV) to α4(IV) chains are present in the basement membrane at the dermal–epidermal junction, with [α1(IV)]2α2(IV) being the predominant heterotrimer.17,21 Type IV collagens chains contain imperfect Gly-X-Y triplets, a feature that confers flexibility to the triple-helical domain of the molecule. A noncollagenous globular domain at the amino-terminal part of the molecule mediates dimer formation, while a short segment at the carboxyl-terminal region of the molecule allows for tetramer assembly, altogether resulting in the so-called “chicken-wire” assembly network for collagens type IV.17 Although skin disorders associated with genetic mutations in type IV collagens have not been described, mutation in the COL4A1 gene has been identified in a family with autosomal dominant porencephaly and infantile hemiparesis,22 and mutations in the COL4A5 gene result in Alport syndrome,23 an X-linked renal disease. Furthermore, autoantibodies recognizing the collagens α3(IV) chain underlie Goodpasture syndrome,23 and autoantibodies targeting the collagens α5(IV) chain are associated with a novel autoimmune disease characterized by subepidermal blisters and renal insufficiency.24
Two further collagens, type VI collagens and type VII collagens, are unique and each has its own distinct molecular structure and supramolecular assembly. Type VI collagens forms specific microfibrils in the dermis, while type VII collagens forms the anchoring fibrils of the dermal–epidermal junction. The microfibril-forming type VI collagens is relatively abundant in a variety of tissues, including skin. In humans, five distinct α chains—α1(VI), α2(VI), α3(VI), α5(VI), and α6(VI)11—have been identified (Table 63-2). The α1(VI), α2(VI), and α3(VI) chains fold into a triple-helical domain of approximately 100 nm in length, with globular domains at both ends (Fig. 63-4). The supramolecular assembly involves the formation of antiparallel dimers, then tetramers, which in turn align in rows, leading to relatively thin microfibrils, independently of the broad collagens fibers.25,26 The microfibrils of type VI collagens may perform an anchoring function by stabilizing the assembly of the broad collagens fibers as well as basement membranes. Studies have now demonstrated that mutations in each of the three type VI collagens genes can lead to different forms of congenital muscular dystrophy, with multiple joint contractures, laxity of distal joints, and characteristic skin involvement.27
Type VII collagens, the major, if not the exclusive, component of the anchoring fibrils at the dermal–epidermal junction, has an unusually long triple-helical region of approximately 450 nm.28 It has only one type of α chain, α1(VII), and the triple-helical collagenous domain is flanked by a large, nonhelical (noncollagenous) domain, NC-1, at the amino terminus and a shorter carboxyl-terminal nonhelical domain, NC-2 (Fig. 63-4). Type VII collagens molecules become organized into anchoring fibrils, which extend from the lower part of the lamina densa to the papillary dermis, through the formation of antiparallel dimers linked through their carboxyl-terminal ends. The large amino-terminal, noncollagenous domains of type VII collagens interact with type IV collagens and laminin 332 components of the dermal–epidermal basement membrane, and it has been suggested that most anchoring fibrils form U-shaped loops that entrap broad dermal collagens fibers consisting of type I and III collagens. Thus, alterations in the expression, structure, or molecular interactions of type VII with other basement membrane components could result in skin fragility. Such a situation is exemplified by dystrophic epidermolysis bullosa (DEB), a group of mechanobullous diseases characterized by blistering of the skin after minor trauma (see Chapter 62). In fact, distinct mutations in the type VII collagens gene, COL7A1, have been demonstrated in more than 500 families with different forms of DEB, and none of the families with DEB studied so far has revealed mutations in genes other than that encoding the α1(VII) polypeptide of type VII collagens.29–31 Furthermore, type VII collagens serves as the autoantigen in the acquired form of epidermolysis bullosa, an autoimmune disorder with circulating antitype VII collagens antibodies (see Chapter 60).
A further class groups the Fibril-Associated Collagens with Interrupted Triple helices, in short FACITs, including types IX, XII, XIV, XVI, XIX, XX, XXI, XXII, and XXVI.11,32 An association with collagens fibers has been demonstrated for several of these collagens, hence the concept that they serve as important molecular bridges for the organization and stability of extracellular matrices. These collagens types form homotrimers with a relatively short triple-helical domain, and 2–4 noncollagenous domains (Fig. 63-4). In the skin, type XII and type XIV collagens associate with the large collagens fibrils of the dermis.
Type XV collagens and Type XVIII collagens are basement membrane-associated collagens. They have been called multiplexins because they contain multiple noncollagenous domains in their collagenous sequences.33 Interest in these collagens stems from the fact that proteolysis liberates fragments endowed with functions different from those of the original intact molecules.33,34
Type XIII, XVII, XXIII, and XXV collagens are transmembrane proteins.35 Their supramolecular assemblies remain unknown, and, except for types XIII and XVII, their presence in skin has not been established. Type XVII collagens (Fig. 63-4) is particularly important for skin physiology and pathology. It is a transmembrane protein anchored in the membrane of basal keratinocytes, with an intracellular domain and a large extracellular domain, or ectodomain, which is a component of the basement membrane at the dermal–epidermal junction. Immunoelectron microscopy has localized type XVII collagens to hemidesmosomes of basal keratinocytes and to the thin anchoring filaments originating from the hemidesmosomes and spanning the lamina lucida toward the lamina densa of the basement membrane.36 The ectodomain of type XVII collagens consists of 15 collagenous domains with the characteristic repeating Gly-X-Y sequences that form triple helices. These collagenous domains are separated by noncollagenous segments of variable sizes, and consequently type XVII collagens is a protein characterized by alternating collagenous and noncollagenous segments.37 The ectodomain colocalizes and interacts with laminin 332, also a component of anchoring filaments36,38 (see also Chapter 53). Type XVII collagens was initially identified as the 180-kDa bullous pemphigoid antigen (BPAG2), which was recognized by circulating autoantibodies in the sera of patients with bullous pemphigoid or herpes gestationis.39 The importance of the type XVII collagens was also shown by mutations in the corresponding gene (COL17A1) that underlie a nonlethal variant of junctional epidermolysis bullosa, generalized atrophic benign epidermolysis bullosa (see Chapter 62). These patients have protracted, lifelong blistering of the skin, atrophic scarring, alopecia, and nail dystrophy.37,40
The collagens genes, like most eukaryotic genes, are large, multiexon genes interrupted at several points by noncoding DNA sequences called introns.41 For expression, the entire gene is transcribed into a high-molecular-weight precursor messenger RNA (mRNA), which undergoes posttranscriptional modifications, such as capping, polyadenylation, and intron splicing to yield a linear, uninterrupted coding sequence with 5′ and 3′ untranslated flanking regions. The mature mRNA is then transported into the cytoplasm and translated to the corresponding polypeptide. Thus, the eukaryotic gene coding for a protein is much larger than would be predicted from the amino acid sequences of the final protein.
With few exceptions, the collagens genes are widely scattered throughout the human genome (see Table 63-2). Knowledge of the precise chromosomal location of the genes coding for collagens in human skin has allowed identification of polymorphic markers within the genes and in the flanking DNA for use in genetic linkage studies. In addition, sophisticated mutation detection strategies, based on scanning of the genes, have led to identification of a large number of mutations in different collagens genes with characteristic phenotypic consequences.
Under physiologic conditions, fibril-forming collagens molecules spontaneously assemble into insoluble fibers.17 This observation presented a logistic problem, because it was difficult to visualize how a collagens molecule could be synthesized inside the cell and then secreted into the extracellular space without premature assembly of the molecules into insoluble fibers. The solution to this problem was found in the demonstration that fibril-forming collagens, and some other collagens, is initially synthesized as a larger precursor molecule, procollagen, which is soluble under physiologic conditions.
The precursor polypeptides of procollagen, so-called pre-proα chains, are synthesized on the ribosomes of the rough endoplasmic reticulum in fibroblasts and related cells (Fig. 63-5). This initial translation product contains an amino-terminal signal (or leader) sequence rich in hydrophobic amino acids. This sequence serves as a signal for attachment of the ribosomes to the membranes of the rough endoplasmic reticulum and vectorial release of the nascent polypeptides into the cisternae of the rough endoplasmic reticulum. During the transmembrane transport of the polypeptides, the signal sequence is enzymatically removed in a reaction catalyzed by signal peptidase, and the polypeptides, termed proα chains, are released inside the lumen of the rough endoplasmic reticulum. For some collagens types, in particular the fibril-forming collagens, proα chains are larger than collagens α chains because they contain additional peptide sequences at both ends of the molecule.
Figure 63-5
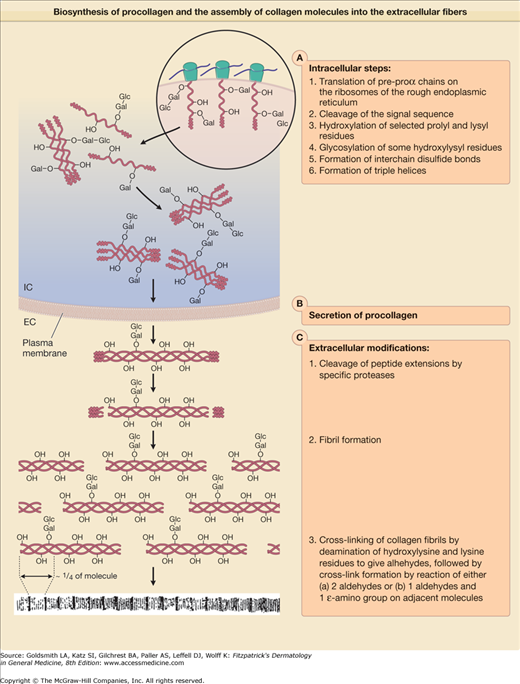
Biosynthesis of procollagens and the assembly of collagens molecules into the extracellular fibers. EC = extracellular; Glc-Gal = glucosylgalactose attached to a hydroxylysyl residue; IC = intracellular; mRNA = messenger RNA; –OH = hydroxyl group of hydroxyproline or hydroxylysine; RER = rough endoplasmic reticulum.
After the assembly of amino acids into pre-proα chains on the ribosomes, the polypeptides undergo several modifications before the completed collagens molecules are deposited into extracellular fibers (Fig. 63-5). Most of these posttranslational modifications are catalyzed by specific enzymes (Table 63-3), and include (1) synthesis of hydroxyproline by hydroxylation of selected prolyl residues; (2) synthesis of hydroxylysine by hydroxylation of selected lysyl residues; (3) attachment of carbohydrates, galactose, or glucosylgalactose, onto certain hydroxylysyl residues; (4) chain association, disulfide bonding, and triple-helix formation; (5) proteolytic conversion of procollagens to collagens; and (6) fiber formation and cross-linking. Current evidence indicates that modification reactions (1)– (4) are intracellular events, whereas proteolytic conversion, fiber formation, and cross-linking probably take place extracellularly (see Fig. 63-5).
Enzymea | Substrate | Product | Co-Factors and Co-Substrates |
|---|---|---|---|
Signal peptidase | Nascent pre-proα chains | Proα chains | None known |
Prolyl-4-hydroxylase | Prolyl residue in X-Pro-Gly sequence in proα chainsb | 4-Hydroxyproline | O2, Fe2+, α-ketoglutarate, ascorbic acid |
Prolyl-3-hydroxylase | Prolyl residue in Pro-Hyp-Gly sequence in proα chainsb | 3-Hydroxyproline | O2, Fe2+, α-ketoglutarate, ascorbic acid |
Lysyl hydroxylase | Lysyl residue in Lys-Gly, Lys-Ser, or Lys-Ala sequence in proα chainsb | Hydroxylysine | O2, Fe2+, α-ketoglutarate, ascorbic acid |
Collagens galactosyl transferase | Hydroxylysine in proα chainsb | Gal-O-hydroxylysine | Mn2+, UDP-galactose |
Collagens glucosyl transferase | Galactosyl-O-hydroxylysine in proα chainsb | Glc-Gal-O-hydroxylysine | Mn2+, UDP-glucose |
Protein disulfide isomerasec | Cysteine residues in the extensions of proα chains | S–S bonds stabilizing the correct protein conformation | Thiols |
Procollagen N-proteinase (ADAMTS-2) | Procollagen or pn-collagens | Pc-collagens or collagensd | Ca2+ |
Procollagen C-proteinase | Procollagen pc-collagens | Pn-collagens or collagensd | Ca2+ |
Lysyl oxidases | Lysyl or hydroxylysyl residue in fibrillar collagens | Aldehyde derivatives of lysine or hydroxylysine | Cu2+, O2 |
A characteristic feature of collagens is the presence of hydroxyproline and hydroxylysine residues.9 Free hydroxyproline and hydroxylysine are not incorporated into nascent polypeptide chains, but result from the hydroxylation of prolyl and lysyl residues, respectively (Fig. 63-6). The hydroxylation reactions are catalyzed by enzymes belonging to the prolyl and lysyl hydroxylase families.9,41–45 These enzymes require molecular oxygen, ferrous iron, α-ketoglutarate, and a reducing agent, such as ascorbate as cosubstrates or cofactors for the reactions (Fig. 63-6 and Table 63-3). Hydroxylation begins while the proα chains are growing on the ribosomes, and it is completed soon after the release of full-length polypeptide chains from the ribosomes (Fig. 63-7). The hydroxyproline in collagens is found in two isomeric forms, trans-3-hydroxy-l-proline, and the most abundant trans-4-hydroxy-l-proline. A critical amount of trans-4-hydroxy-l-proline is a prerequisite for the folding of α chains into the triple helix, the conformation required for secretion of procollagens molecules out of the cells. Because triple-helix formation takes place in the cisternae of the rough endoplasmic reticulum, and because prolyl hydroxylases do not hydroxylate prolyl residues if the collagens substrate is in a triple-helical conformation, hydroxyproline formation must be completed before the procollagens molecules leave this cellular compartment. Thus, in the absence of hydroxyproline, the critical triple-helical structure of collagens would not form under physiologic conditions, and no functional collagens fibers would appear in the extracellular space. Also, because prolyl hydroxylase requires a reducing agent, such as ascorbate, for its activity, ascorbic acid deficiency leads to a decreased formation of collagens fibers. This explains some of the clinical manifestations in scurvy, such as poor wound healing and decreased tensile strength of the connective tissues.46 An analogous situation may exist in tissues with hypoxia, because molecular oxygen is a specific requirement for the formation of hydroxyl groups in hydroxyproline. Studies in animal models demonstrate that wound healing is relatively poor under hypobaric conditions, and in such situations the low oxygen levels may limit the synthesis of hydroxyproline.47 This observation may also explain the decreased healing tendency of wounds and ulcers in peripheral tissues that are anoxic due to relatively poor blood supply. It may also relate to the recent appreciation that prolyl hydroxylases are genuine oxygen sensors. In addition to the prolyl-4-hydroxylase playing a critical role in the hydroxylation of prolyl residues on nascent collagens polypeptide chains, there are additional prolyl-4-hydroxylase isoforms that are responsible for the hydroxylation of two proline residues that earmarks the α subunit of the hypoxia-inducible transcription factor (the master regulator of hypoxia-inducible genes) for proteasomal degradation.44,48,49
Figure 63-6
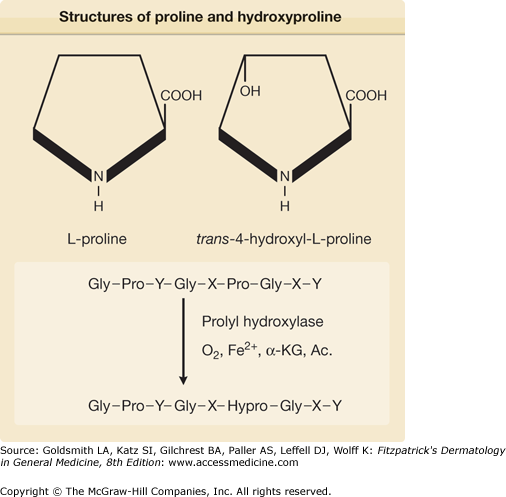
Structures of proline (Pro) and hydroxyproline (Hypro), and schematic representation of the enzymatic hydroxylation of prolyl residues in the Y position of the repeating Gly-X-Y sequence of collagens polypeptides. Ac = ascorbic acid; Gly = glycine; α-KG = α-ketoglutarate. (From Uitto J, Prockop DJ: Inhibition of collagens accumulation by proline analogs: The mechanism of their action. In: Collagens Metabolism in the Liver, edited by H Popper, K Becker. New York, Stratton Intercontinental, 1975, p. 139, with permission.)
Figure 63-7
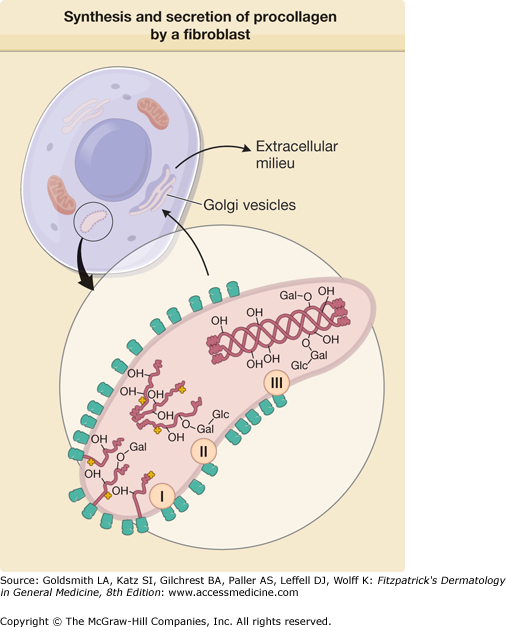
Representation of the synthesis and secretion of procollagen by a fibroblast. The enlarged area demonstrates events taking place in the rough endoplasmic reticulum of the cells during the synthesis of procollagen. In the first stage (I), the polypeptide chains of procollagen are synthesized on the membrane-bound ribosomes, and the nascent chains are fed into the cisternae of the rough endoplasmic reticulum. Hydroxylation of prolyl and lysyl residues and glycosylation of hydroxylysyl residues are initiated on the growing polypeptide chains, and these reactions are completed soon after the release of full-size chains from ribosomes (II). Three proα chains are linked together by the formation of interchain disulfide bonds, and the collagenous portions of the polypeptides assume a triple-helical conformation (III). The procollagen molecules are then transferred from the rough endoplasmic reticulum to Golgi vesicles and are secreted from these vesicles into the extracellular milieu. –OH = hydroxyl groups of hydroxyproline and hydroxylysine; –Gal = galactosyl residue attached to hydroxylysine; –Gal-Glc = glucosylgalactosyl residue attached to hydroxylysine in O-glycosidic linkage; the cloverleaf-like structures signify the hydroxylating and glycosylating enzymes. (After Prockop DJ et al: Intracellular steps in the biosynthesis of collagens. In: Biochemistry of Collagens, edited by GN Ramachandran, AH Reddi. New York, Plenum, 1976, p. 163, with permission.)
Lysyl hydroxylation is of critical importance because hydroxylysyl residues serve either as an attachment site for galactosyl and glucosylgalactosyl residues during the intracellular synthesis of procollagen, or to the formation of cross-links that stabilize the collagens matrix in the extracellular space.9,45,50 The intracellular modification of lysyl residues is a sequential event, with first hydroxylation of lysyl residues, followed by the attachment of galactosyl residue to the hydroxyl group of hydroxylysine with an O-glycosidic bond and then a glucose moiety is attached to some of the galactosyl residues (see Figs. 63-5 and 63-7). Therefore, the synthesis of hydroxylysine is a prerequisite for the glycosylation of collagens (see Table 63-3). Three lysyl hydroxylase isoforms exist, and one of them, the lysyl-3-hydroxylase (LH3), displays three activities: (1) lysyl hydroxylase, (2) galactosyltransferase, and (3) glucosyltransfrase activities.45 The glycosylation reactions use uridine diphosphate sugars as a source of the carbohydrate and require Mn2+ as a cofactor. In addition to the glycosylation of hydroxylysyl residues in the triple-helical portion of the molecule, the nonhelical extensions contain complex carbohydrates, consisting mainly of mannose. Deficiency of lysyl hydroxylase has been identified in patients with the scoliotic form (type VI) of Ehlers–Danlos syndrome, characterized by hyperextensible skin, loose-jointedness, severe kyphoscoliosis, and ocular fragility. A connective tissue disorder caused by mutations of the lysyl hydroxylase 3 gene has been recently identified in humans. The disorder is associated with abnormalities in several organs, including skin, and the phenotype has features that overlap with a number of known collagens disorders.51
A critical step in the intracellular biosynthesis of procollagens is the association of three proα chains and subsequent folding of the collagens portion of the polypeptides into a triple helix (see Fig. 63-7). The noncollagenous peptide extensions on the individual proα chains assume globular conformations soon after their translation, and this conformation contains the specific information that directs the correct association of the three proα chains. Such a mechanism might explain the association of proα1 and proα2 chains in a proper 2:1 ratio during the synthesis of type I procollagen. It would also explain the rapid and efficient association of the proα chains and folding of the molecule into the triple helix. The association of the extensions at the carboxyl-terminal ends of the polypeptide chains appears to facilitate folding of the molecules into the triple helix, perhaps by providing a nucleation site from which the formation of the triple helix is propagated throughout the collagenous portion of the molecule.
After secretion into the extracellular space, procollagens molecules are converted to collagens by limited proteolysis, which removes the extension peptides on the molecule.52 The conversion of type I procollagens to collagens is catalyzed by two specific enzymes, (1) procollagens N-proteinase and (2) procollagens C-proteinase, that separately remove the amino-terminal and carboxyl-terminal extensions, respectively (see Table 63-3). Furthermore, the N-proteinase catalyzing the conversion of type III procollagens to collagens is a separate, specific enzyme.
The N-proteinase capable of cleaving type I procollagens belongs to the ADAMTS (a disintegrin and metalloprotease with thrombospondin motifs) family of extracellular proteases, and is called ADAMTS-2.53 The activity of this enzyme is dependent on the native conformation of the amino-terminal propeptides in procollagen, because it does not catalyze the removal of the extension peptides from isolated proα chains. Furthermore, partially purified N-proteinase is inhibited by metal chelators, which suggests a requirement for divalent cations.
C-proteinase is required for the removal of the carboxyl-terminal extension from type I, II, III, and V procollagen, which allows the fully processed molecules to form functional fibers.52 Cloning of type I procollagens C-proteinase revealed that it is identical to bone morphogenic protein-1, a metalloprotease implicated in pattern formation during development of diverse organisms and also capable of inducing ectopic bone formation.54 The activity of C-proteinase/bone morphogenic protein-1 is stimulated by procollagens C-proteinase enhancers, glycoproteins that bind to the C-terminal propeptide of type I procollagen.55 Thus, the conversion of procollagens to collagens is a complex, carefully controlled process, and lack of removal of either the amino- or the carboxyl-terminal extensions results in impaired tensile strength of collagens fibers in the skin. Specifically, deficiency in the removal of the amino-terminal propeptide of type I collagens in vivo causes dermatosparaxis, a disease of fragile skin, originally recognized in various animal species, and more recently recognized in humans.56 Specifically, the human counterpart is the dermatosparaxis type of Ehlers–Danlos syndrome (type VIIc), caused by deficiency in N-proteinase activity. It should be noted that a disease with similar phenotype, the arthrochalasia type of Ehlers–Danlos syndrome (types VIIa and VIIb), can be caused by mutations in the type I collagens genes (COL1A1 and COL1A2, respectively) at the cleavage site for the N-proteinase.57
Collagens assembly is best defined for fibrillar collagens. After removal of the extension peptides in the extracellular space, the collagens molecules spontaneously align to form fibers. However, these fibers do not attain the necessary tensile strength until the molecules are linked together by specific covalent bonds known as cross-links.58 The most common forms of cross-links in collagens are derived from lysine or hydroxylysine. The first step in the cross-linking of collagens is the enzymatic conversion of some of the lysyl and hydroxylysyl residues to the corresponding aldehyde derivatives by removal of the ϵ-amino groups (see Figs. 63-5 and 63-7). The aldehydes then form cross-links by two kinds of reactions. One reaction involves condensation of an aldehyde with an ϵ-amino group still present in another unmodified lysine or hydroxylysine to form a Schiff base-type of covalent cross-link. The second type of reaction is an aldol condensation between two aldehydes. In addition to these cross-links, collagens contains several more complex cross-links that also involve lysyl or hydroxylysyl residues. The lysine- and hydroxylysine-derived cross-links can be either intramolecular, occurring between two adjacent α chains in the same collagens molecule, or intermolecular, stabilizing the alignment of neighboring collagens molecules along microfibril structures.
The first step in collagens cross-linking, the oxidative deamination of certain lysyl and hydroxylysyl residues, is catalyzed by lysyl oxidase. This enzyme requires copper as a cofactor, and its activity is readily inhibited by nitriles, such as β-aminopropionitrile, which produce lathyrism in animals. Because the cross-links of collagens provide the tensile strength required in certain tissues, a defect in the formation of these covalent bonds can lead to a disturbance in connective tissue function. An example is occipital horn syndrome (previously known as Ehlers–Danlos syndrome type IX), which results from reduced lysyl oxidase activity (see Chapter 137). The primary defect resides in perturbed copper metabolism caused by mutations in a copper transport enzyme protein, an adenosine triphosphatase encoded by the gene MNK-1 that is also involved in Menkes syndrome.59 As a result, serum copper levels are reduced, which leads to reduced lysyl oxidase activity.
A major question in ECM biology concerns the mechanisms that control the deposition of collagens and other ECM proteins in tissues. Such control must exist in vivo, because the consequences of uncontrolled ECM accumulation are demonstrated by diseases such as progressive systemic sclerosis (see Chapter 157) and various other fibrotic conditions.60,61
The accumulation of ECM in tissues can be controlled at several different levels of biosynthesis and degradation. Several observations suggest that an important control mechanism acts at the level of mRNA formation through regulation of the transcriptional activity of gene expression.62,63 The transcriptional regulation of gene expression involves both cis-acting elements and trans-acting factors. The cis-acting elements are nucleotide sequences in the promoter region of the gene that serve as binding sites for trans-acting cellular proteins, which can upregulate or downregulate the transcriptional promoter activity. Some of the trans-acting factors are nuclear receptors, such as the retinoic acid receptors (RAR and RXR)49 that form a complex with the ligand (a retinoid) and then bind to the retinoic acid-responsive elements (RARE) in the target gene. Retinoids, such as all-trans-retinoic acid, modulate type I collagens gene expression both in vitro and in vivo.64 In particular, quiescent nonproliferating cells can be stimulated by retinoic acid to activate collagens gene expression. This may have relevance to the elevated collagens synthesis observed in photodamaged dermis after topical application of all-trans-retinoic acid (see Chapter 109).
Diverse growth factors, cytokines, and chemokines are released into the extracellular space from different types of cells, such as fibroblasts, keratinocytes, and endothelial and inflammatory cells. By both paracrine and autocrine mechanisms these factors elicit intracellular signaling pathways acting on the transcriptional and/or translational levels. One of the most powerful regulators of collagens and ECM production is transforming growth factor β (TGF-β), a member of a family of growth factors involved in many physiological and pathological conditions.65 Interestingly, TGF-β can induce the expression of other growth factors, in particular the expression of connective tissue growth factor in fibroblasts. Also, cytokines such as platelet-derived growth factor, interleukin-1 and -4, and chemokines, such as monocyte chemotactic protein-1 and -3, are thought to induce collagens production.66
In the extracellular space, most collagens form polymers described above, which in turn interact with other proteins of the ECM through multiple protein–protein interactions. It gives rise to large multimolecular assemblies and to the organization of the ECM proteins into specific networks.17 In addition, many growth factors and cytokines specifically interact with ECM proteins, and they are therefore stored in the networks. There is a continuous remodeling of the multimolecular assemblies, at a rate that can be significantly enhanced during development, wound healing or in certain disease processes. This remodeling results in the release of a large variety of biologically active peptides, including growth factors.34
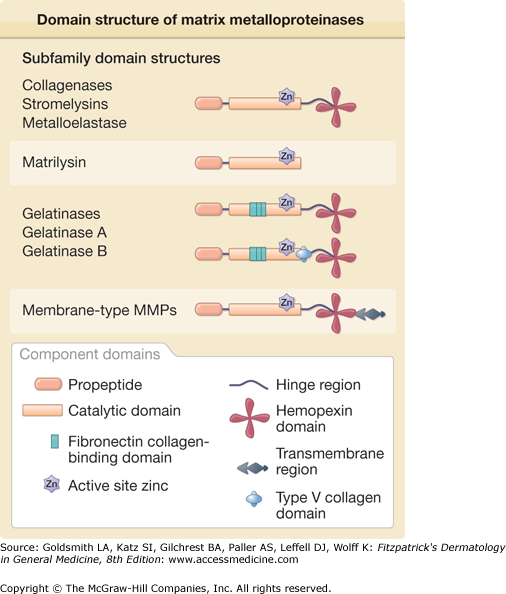
The remodeling of the ECM is a cooperative multistep process involving a variety of molecular pathways and mechanisms. The initial step depends on the presence of proteinases capable of initiating the degradation of the ECM proteins. These enzymes comprise the matrix metalloproteinase (MMP) family,67–71 which includes collagenases, gelatinases, stromelysins, matrilysins, metalloelastase, enamelysin, and the membrane-type MMPs (Fig. 63-8 and Table 63-4). Interstitial collagenase (MMP-1) was the first enzyme of the MMP family to be discovered and was defined by its ability to break down triple-helical collagens that is resistant to most proteases.72 Human skin collagenase was initially isolated in active form from the culture medium of skin explants and subsequently as a proenzyme from monolayer fibroblast cultures.73 Other cell types present in skin, including keratinocytes, endothelial cells, monocytes, and macrophages, express an identical enzyme. Like the other MMPs, MMP-1 contains intrinsic zinc in the active site and requires calcium for its activity and thermostabilization.
Enzyme | MMP Number | Alternate Name | Proenzyme Molecular Weight | Known Matrix Substrates |
|---|---|---|---|---|
Interstitial collagenase | MMP-1 | Type I collagenase | 52,000 | Collagens I, II, III, VII, VIII, X, entactin, tenascin, aggrecan, denatured collagens, interleukin-1β, myelin basic protein, L-selectin |
Neutrophil collagenase | MMP-8 | — | 75,000 | Collagens I, II, III, V, VII, VIII, X, gelatin, aggrecan, fibronectin |
Collagenase-3 | MMP-13 | — | 52,000 | Collagens I, II, IV, IX, X, XIV, aggrecan |
Gelatinase A | MMP-2 | 72-kDa type IV collagenase | 72,000 | Denatured collagens, collagens IV, V, VII, X, XI, XIV; collagens I, species dependent; elastin; fibronectin; laminin; aggrecan; myelin basic protein |
Gelatinase B | MMP-9 | 92-kDa type IV collagenase | 92,000 | Denatured collagens, collagens IV, V, VII, X, XIV, elastin, entactin, aggrecan, fibronectin, osteonectin, interleukin-1β, plasminogen, myelin basic protein |
Stromelysin-1 | MMP-3 | Proteoglycanase | 57,000 | Proteoglycan core protein, laminin, fibronectin, collagens I, IV, V, IX, X, XI, gelatin, elastin, tenascin, aggrecan, myelin basic protein, entactin, decorin, osteonectin |
Stromelysin-2 | MMP-10 | Transin-2 | 55,000 | Proteoglycan core protein, collagens III, IV, V, laminin, fibronectin, elastin, aggrecan |
Stromelysin-3 | MMP-11 | — | 61,000 | α1-proteinase inhibitor |
Matrilysin | MMP-7 | PUMP, matrilysin-1 | 28,000 | Collagens IV, denatured collagens, laminin, fibronectin, elastin, aggrecan, tenascin, myelin basic protein |
Matrilysin-2 | MMP-26 | Endometase | 28,000 | Gelatin, α1-proteinase inhibitor |
Membrane-type MMP-1 | MMP-14 | MTI-MMP | 63,000 | Progelatinase A, denatured collagens, fibronectin, laminin, vitronectin, entactin, proteoglycans |
Membrane-type MMP-2 | MMP-15 | MT2-MMP | 72,000 | Progelatinase A |
Membrane-type MMP-3 | MMP-16 | MT3-MMP | 64,000 | Progelatinase A |
Membrane-type MMP-4 | MMP-17 | MT4-MMP | 70,000 | Unknown |
Membrane-type MMP-5 | MMP-24 | MT5-MMP | 73,000 | Progelatinase A |
Membrane-type MMP-6 | MMP-25 | MT6-MMP | 63,000 | Unknown |
Metalloelastase | MMP-12 | — | 54,000 | Elastin, collagens IV, vitronectin, plasminogen, laminin, entactin, fibrinogen, fibrin, fibronectin |
Enamelysin | MMP-20 | — | 54,000 | Amelogenin, aggrecan |
MMP-19 | MMP-19 | RASI-1 | 57,000 | Gelatin, aggrecan, fibronectin |
MMP-21 | MMP-21 | — | Unknown | Unknown |
MMP-22 | MMP-22 | — | Unknown | Unknown |
MMP-23 | MMP-23 | — | 44,000 | Unknown |
Epilysin | MMP-28 | — | 56,000 | Unknown |
MMP-1 degrades some of the collagens present in skin, such as types I, III, and VII, but not others like types IV and V.74 At physiologic pH and temperature, the enzyme catalyzes a single cleavage three-quarter of the distance from the amino terminus in each of the three α chains comprising the triple-helical, native collagens monomer. Specifically, MMP-1 cleaves the α1(I) chain at a Gly-Ile (glycine-isoleucine) bond (residues 775 and 776) and the α2(I) chain at a Gly-Leu (glycine-leucine) bond in the homologous region. Ten other Gly-Ile or Gly-Leu bonds within the triple-helical domain of interstitial collagens are not cleaved, which suggests that the local conformation of the collagenase cleavage site is a major factor in determining substrate specificity. Indeed, MMP-1 catalyzes multiple cleavages in the denatured chains of all collagens types at Gly-Ile and Gly-Leu bonds. Thus, it appears that the triple-helical conformation of native collagens can drastically alter MMP-1 ability to catalyze cleavages that would be permissible from primary sequence consideration alone.
Human neutrophil interstitial collagenase (MMP-8) attacks collagens at the same site, as does human MMP-1, to produce the characteristic three-quarter/one-quarter collagens fragments. Human MMP-8 is highly homologous to MMP-1, but has a higher degree of glycosylation. It is not immediately secreted but is stored in neutrophil granules and released on stimulation. Although both fibroblast (MMP-1) and neutrophil (MMP-8) collagenases have similar affinities for type I and III collagens, fibroblast collagenase degrades soluble type III collagens with a higher turnover rate, whereas neutrophil collagenase degrades soluble type I collagens more rapidly. For both enzymes, the differences in specificity against monomeric collagens are largely abolished when the substrates are reconstituted into the insoluble fibrillar forms found in tissues.75
Important mechanistic information on collagens degradation has recently emerged, and specifically, it has been shown75 that activated collagenase (MMP-1) moves processively on collagens fibrils in which the mechanism of movement is a biased diffusion with the biased component of the motion depending on the proteolysis of collagens and not on ATP hydrolysis. Most importantly, the closely related MMPs, MMP-2 (gelatinase) MMP-9, and the extracellular portion of the membrane-associated metalloproteinase MT1–MMP, can also diffuse on collagens fibrils. These studies also show that a collagenolytic MT1–MMP complex exists at the cell surface. The MT1–MMP complex anchored at the cell surface has profound implications concerning the generation of the forces required for cell locomotion, not only in the normal physiologic processes of tissue remodeling but also in pathologic processes, such as tumor invasion.
Collagenase-3 (MMP-13), first cloned from breast carcinoma, is homologous to rodent collagenase.69
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


