Abstract
As the outer barrier to the environment, the skin is the organ most challenged by a range of external stress factors that can lead to cellular damage and barrier disruption. The skin has developed complex mechanisms to protect itself following injuries, including rapidly restoring tissue integrity and function. Over the past two decades, considerable insights have been gained into the cellular and molecular pathways regulating the wound healing response. Multiple strategies have been developed to try to enhance endogenous repair mechanisms, both in experimental systems and by clinical approaches. However, the precise mechanisms that lead to a failure of skin repair and chronic, recalcitrant skin ulcers are still poorly understood. Thus, poor wound healing that can follow trauma and surgical procedures or is associated with chronic disorders (e.g. venous hypertension, atherosclerosis, diabetes mellitus) affects millions of people worldwide. Chronic skin ulcers represent a common diagnostic and therapeutic challenge. In order to offer new effective therapies, we need to better understand the heterogeneity and the complex molecular pathophysiology of chronic wounds.
Keywords
wound healing, impaired wound healing, mechanisms of wound healing, pathophysiology of impaired wound healing, chronic ulcers, pathophysiology of chronic ulcers, skin ulcers, cutaneous ulcers, stem cells
- ▪
Wound healing is a complex and dynamic biologic process, consisting of three consecutive phases: inflammation, tissue formation, and tissue remodeling
- ▪
Effective wound healing requires synchronization of cell–cell and cell–matrix interactions as well as an interplay of cytokines
- ▪
Extracellular matrix proteins are multifunctional molecules that bind directly to cell surface receptors (e.g. integrins) and influence the effects of growth factors on cells (e.g. TGF-β)
- ▪
In children and adults, the cutaneous wound healing response represents a reparative process that can lead to fibrosis (scar), whereas injured fetal tissue can heal completely without fibrosis, in a process resembling regeneration
- ▪
A number of medical disorders (e.g. venous hypertension, atherosclerosis, diabetes mellitus) as well as more local factors (e.g. pressure, infection) are associated with chronic non-healing wounds
Introduction
Restoration of skin integrity and homeostasis following injury is a fundamental process that ensures survival. The primary goal of the wound healing response is to re-establish a functional skin barrier as quickly as possible. Ideally, the wound healing response would lead to complete regeneration of skin tissue and its adnexal structures, with complete restoration of original skin function and morphology. Unfortunately, this is often not the final result of wound repair and the quality of newly formed skin tissue varies considerably.
Regeneration Versus Repair
In children and adults, the wound healing response characteristically leads to fibrosis, i.e. scar formation. In addition, adnexal structures (e.g. hair follicles, sweat and sebaceous glands) as well as components of the dermal extracellular matrix may fail to regenerate, resulting in a loss of normal skin function and impaired morphology. Because it is less than ideal, this type of wound healing is sometimes referred to as a reparative process . In contrast, during embryogenesis injured fetal skin can heal completely without fibrosis. Thus, the process resembles regeneration .
The underlying mechanisms that determine whether tissue regeneration versus tissue repair will occur still remain a mystery. Insights will hopefully come from experimental studies in other multicellular organisms (e.g. amphibians, fish) that retain throughout their adult life the capability to regenerate tissues after injury, as well as from studies of wound repair in fetal skin. In the future, these insights could lead to methods for transforming repair into regeneration and as a result, provide novel therapies that allow wound healing with minimal scarring.
Effect of Immune Response on Wound Healing
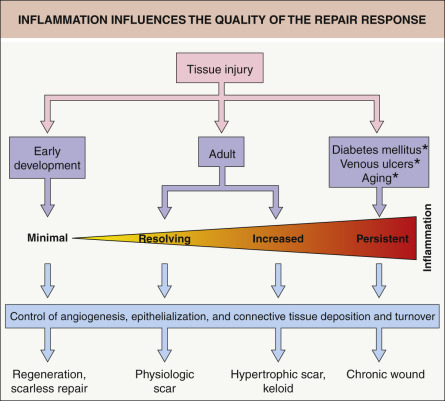
The immune system, including both innate and adaptive arms of the immune response, plays a critical role in wound healing. By influencing multiple repair mechanisms (e.g. angiogenesis, connective tissue deposition, epithelialization), inflammation has an impact on all stages of the repair response and ultimately the extent of scarring ( Fig. 141.1 ). In a number of models, there appears to be an inverse correlation between the intensity of the inflammatory immune response and the ability to undergo regeneration, with inappropriate immune reactions leading to tissue damage and impairment of tissue repair. However, this paradigm has recently been challenged by studies in several model organisms in which inflammatory signals were found to be crucial for promoting timely repair and for inducing fundamental processes involved in regeneration . Of note, these novel and unexpected findings emerged from investigations that employed well-established models within the field of tissue regeneration (not immunology).
Mechanisms of Tissue Repair and Regeneration Are Evolutionarily Conserved
Amphibians
Among the vertebrates, amphibians and fish are exceptional in their capacity to regenerate anatomically complete and fully functional tissues and organs during adulthood. In particular, urodele amphibians (newts and salamanders) can regenerate a range of organs and tissues . Cellular and molecular studies have focused primarily on limb regeneration after amputation, and recently such studies in adult salamanders demonstrated that the immediate influx of macrophages after tissue damage (preceding blastema formation) was an essential component of regeneration .
Zebrafish
Zebrafish also represent a traditional and valuable model for the study of regeneration. Adult zebrafish maintain regenerative capacity, not only after caudal fin amputation, but also following skin injury . Of interest, in both fin and skin regeneration there is an infiltration of myeloid inflammatory cells, suggesting that in principle regeneration can occur in the presence of inflammatory signals. Thus, by examining skin wound healing in zebrafish it may be possible to distinguish between beneficial and harmful inflammatory mediators and how they influence scar formation.
Mammals
The effects of inflammation on regeneration and repair have also been studied in mammals, primarily in mice, but also in humans. In addition to transgenic mouse models, refined mouse models that permit inducible and time-restricted depletion of specific immune cells have unraveled specific and critical functions of individual immune cell lineages in skin repair. For example, they provided evidence that, besides their central role in clearing cell detritus and microorganisms, macrophages exert distinct functions during the various phases of repair . Notably, macrophages recruited immediately after injury are essential for the induction of vascular sprouts and angiogenesis . Also, as the wound begins to contract, there is activation of focal adhesion kinase/extracellular signal-regulated kinase (FAK-ERK) within fibroblasts which leads to release of chemokine ligand 2 (CCL2), one of the prime chemokines known to drive monocyte/macrophage recruitment . Thus, an intimate interaction between macrophages and fibroblasts is envisioned.
In the presence of a functional immune system, higher vertebrates, including rodents and humans, can undergo post-amputation fingertip regeneration . Previously it was thought that the blastema, an undifferentiated pluripotent cell population presumably derived from mature cells via dedifferentiation, was responsible for mouse distal digit regeneration. However, recent genetic fate mapping and clonal analysis of individual cells revealed that a wide range of lineage-restricted tissue stem/progenitor cells contributed to restoration of the mouse distal digit, rather than pluripotent blastema cells . In humans, conservatively managed amputation injuries in children (but not adults) can be followed by restoration of the contour, fingerprints, and normal sensation and function of the digit, with minimal scarring . While the molecular mechanisms underlying this phenomenon are not yet understood, investigation of conserved mechanisms in more easily trackable non-human models will hopefully allow extrapolation to humans.
Clinical Implications
The underlying etiology of chronic, non-healing ulcers is multifactorial (see Ch. 105 ). It includes hypoxia due to vascular disease or sustained pressure, infection, and persistent inflammation . How the latter impairs the healing response is still not well understood, but increased protease activity (e.g. metalloproteinases [MMPs], serine proteinases) and generation of reactive oxygen species are thought to play a major role. Unravelling the underlying molecular pathophysiology of chronic wounds in humans will clearly involve studies of both regeneration and repair.
Impact of Wound Depth on Wound Healing
Skin wounds are often categorized according to their depth ( Fig. 141.2 ). Defects that affect only the epidermis (or a portion of the epidermis) are referred to as erosions . When the wound extends into the dermis, it is termed an ulceration . In “partial-thickness” wounds, the epidermis and a portion of the dermis are missing, with the ulcer extending into the mid dermis, but the adnexal structures remain. Full-thickness wounds, on the other hand, involve the entire dermis and extend into the subcutaneous fat. As a result, adnexal structures are lost as a source of keratinocytes for re-epithelialization.
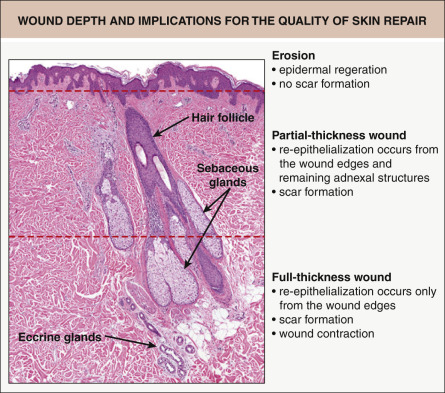
The depth of a skin injury clearly affects the capacity of the skin to repair or regenerate. When erosions heal, there is regeneration of the entire epidermis, without scarring (see Fig. 141.2 ). Ulcerations, however, heal via a reparative process and are associated with scar formation. In partial-thickness wounds, the preserved adnexal structures serve as a source of epithelial cells to repopulate the epidermis. Epithelia from these structures, as well as from the wound edge, migrate across the wound surface to provide coverage. In contrast, because adnexal structures are lost in full-thickness wounds (see above), re-epithelialization can only occur from the wound edges.
Healing of full-thickness wounds includes, to some extent, contraction. While there is minimal contraction in partial-thickness wounds, the reason for this difference is not clear. Contraction may be mediated by mechanical or biologic factors, e.g. the differentiation of fibroblasts into myofibroblasts. Recent studies in mice suggest that the contributions of different stem cell sources for wound fibroblasts may determine the outcome of the repair response. These include a superficial population critical for hair development and a deeper population that forms the lower dermis and early on may provide a source of locally-derived wound fibroblasts that can develop into myofibroblasts .
During contraction, the wound area decreases via centripetal movement of pre-existing tissue, not the formation of new tissue. Unfortunately, contraction of wounds may result in cosmetically disfiguring contractures. However, the contraction of wounds occurs in predictable directions (in relation to “skin tension lines”), and in order to direct the contracture, surgical incisions should be placed, if possible, parallel to these skin tension lines. Also, an argument in favor of the use of full-thickness skin grafts is to prevent wound contraction and subsequent contracture (see Ch. 148 ).
Primary Versus Second Intention Healing
When an acute wound, such as one resulting from a surgical excision, is allowed to heal on its own, this is termed second (secondary) intention healing (see Ch. 146 ). In primary intention healing, closure of the wound is accomplished by approximating the wound edges; the latter includes side-to-side closures, flaps, and grafts. Both primary and second intention forms of wound healing proceed through the three phases of healing.
In second intention healing, the time interval until complete re-epithelialization depends upon several factors. These include wound depth, anatomic location (e.g. facial wounds heal faster than acral wounds), any secondary infection, vascular supply, and geometric shape (for a given area, a more narrow diameter will heal faster). For smaller wounds, especially those that are partial-thickness, primary and second intention healing can produce similar cosmetic results. Once wounds have a diameter >8 mm, primary intention healing leads to better cosmetic results. Tertiary intention healing refers to wounds that are closed with the goal of primary intention healing, but dehiscence occurs and the wound is then allowed to heal by second intention.
The decision to choose primary versus second intention wound healing depends on a number of variables, including a patient’s overall medical status and cosmetic concerns, as well as the anatomic location, depth and size of the wound, and whether it is located on a convex or concave surface. In addition to comorbidities such as atherosclerosis, venous hypertension and diabetes mellitus, the risk of infection (which may be increased in open wounds) and the patient’s preference also influence the decision.
Skin Repair – Cellular and Molecular Aspects
In most mammalian organ systems, the repair response involves a complex and dynamic interplay of numerous cell types, including cells that reside within the tissue (e.g. keratinocytes, endothelial cells, fibroblasts) and hematopoietic cells recruited to the site of tissue damage. Three sequential phases of wound repair have been delineated – inflammatory, proliferative, and remodeling (maturation) . This section discusses the associated cellular and molecular events.
Three Phases of Wound Healing
Tissue injury causes leakage of blood constituents into the wound site and release of vasoactive factors ( Table 141.1 ), resulting in the activation of the clotting cascade. Clotted blood provides a matrix that allows for cell adhesion and migration. Not only do platelets trapped within the clot play an important role in hemostasis, they also represent a rich source of growth factors (e.g. PDGF) and proinflammatory cytokines that mediate the recruitment of inflammatory cells and fibroblasts into the wound site ( Fig. 141.3 ). The early inflammatory phase of repair is characterized by local activation of innate immune functions and chemoattraction, both of which lead to an early influx of polymorphonuclear leukocytes (PMNs; neutrophils). This is followed by invasion of blood monocytes, which differentiate into tissue macrophages .
| CHEMICAL MEDIATORS OF INFLAMMATION THAT PLAY A ROLE IN WOUND HEALING | |
|---|---|
| Chemical mediator | Action |
| Histamine | Increased vascular permeability |
| Serotonin | Stimulation of fibroblast proliferation Cross-linking of collagen molecules |
| Kinins | Increased vascular permeability |
| Prostaglandins | Increased vascular permeability |
| Sensitize pain receptors | |
| Increased synthesis of GAGs | |
| Complement | Increased vascular permeability |
| Increased phagocytosis | |
| Enhanced bacterial lysis | |
| Mast cell and basophil activation | |
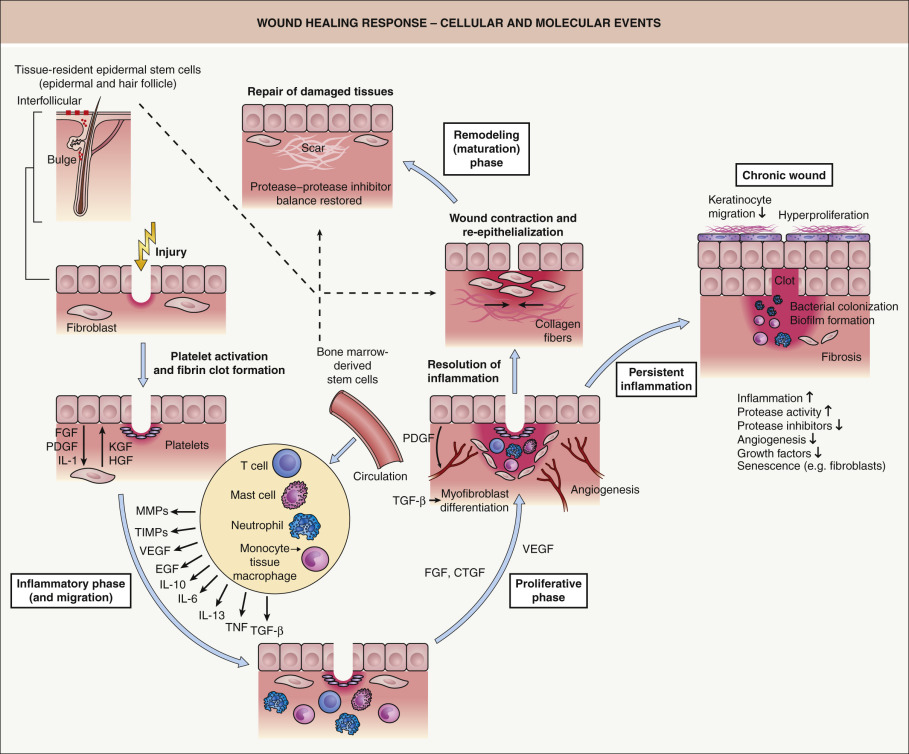
While one function of the inflammatory infiltrate is to combat invading microbes, another equally important function is the release of cytokines and growth factors (e.g. IL-1, IL-6, vascular endothelial growth factor [VEGF], tumor necrosis factor [TNF], TGF-β; Table 141.2 ), which are critical for the initiation of the second proliferative phase of skin repair (see Fig. 141.3 ). During this phase of tissue formation, newly formed granulation tissue, consisting primarily of invading macrophages and fibroblasts as well as endothelial cells (as vessel precursors), covers and fills the wound area. Fibrin, fibronectin, vitronectin, type III collagen, and tenascin are components of the provisional extracellular wound matrix which facilitate cell adhesion, migration and proliferation. At the wound edge, epidermal–mesenchymal interactions stimulate keratinocyte proliferation and migration, leading to re-epithelialization .
| GROWTH FACTORS INVOLVED IN WOUND HEALING | |||
|---|---|---|---|
| Growth factor | Abbreviation | Source | Functions |
| Platelet-derived growth factor | PDGF | Platelets, keratinocytes, fibroblasts, endothelial cells, perivascular cells | Fibroblast proliferation, chemotaxis & collagen metabolism; angiogenesis |
| Transforming growth factor-β | TGF-β | Platelets, keratinocytes, fibroblasts, endothelial cells, macrophages | Fibroblast proliferation, chemotaxis & collagen metabolism; angiogenesis; immunomodulation |
| Transforming growth factor-α | TGF-α | Platelets, keratinocytes | Keratinocyte proliferation & migration |
| Epidermal growth factor | EGF | Platelets | Keratinocyte proliferation & migration |
| Interleukins | IL-1, IL-10 | Leukocytes, keratinocytes | Fibroblast proliferation; proinflammatory (IL-1); anti-inflammatory (IL-10) |
| Tumor necrosis factor | TNF | Leukocytes, keratinocytes | Promotes inflammation |
| Connective tissue growth factor | CTGF | Fibroblasts, endothelial cells | Fibroblast proliferation, chemotaxis & collagen metabolism |
| Fibroblast growth factor | FGF | Keratinocytes, macrophages | Fibroblast & epithelial cell proliferation; matrix deposition, wound contraction; angiogenesis |
| Keratinocyte growth factor | KGF | Fibroblasts | Keratinocyte proliferation |
| Insulin-like growth factor 1 | IGF-1 | Fibroblasts | Keratinocyte proliferation & differentiation |
| Hepatocyte growth factor | HGF | Fibroblasts, macrophages | Keratinocyte proliferation |
| Vascular endothelial growth factor | VEGF | Platelets, keratinocytes, macrophages, neutrophils | Angiogenesis; vascular permeability; macrophage chemotaxis |
Upon completion of epithelialization, cell proliferation and neovascularization cease, scar tissue forms and the wound enters the remodeling ( maturation ) phase, which lasts for several months. This last phase is characterized by a balance between the synthesis of new components of the scar matrix and their degradation by proteases. This balance determines whether there is normal versus abnormal scar formation (e.g. atrophic scars, hypertrophic scars, keloids). The mechanisms underlying granulation tissue regression and its transformation into scar tissue during this phase are largely unknown. Typically, there is also regression of vascular structures, transformation of fibroblasts into myofibroblasts, substitution of provisional extracellular matrix (ECM) with a permanent collagenous matrix, and a final resolution of the inflammatory response. However, if the inflammatory response persists, a disturbed healing response may result, leading to chronic skin ulcers (see Fig. 141.3 ). The mechanisms that direct resolution of the inflammatory response are not precisely understood, but most likely involve increased production of anti-inflammatory mediators, active suppression of proinflammatory factors, normalization of microvascular permeability, induction of apoptosis of inflammatory cells, and epithelialization.
Cellular Components
Keratinocytes
Within hours after injury, keratinocytes at the wound edge are activated and undergo marked phenotypic and functional alterations, a fundamental change required for re-epithelialization of the wound bed ( Fig. 141.4 ). In order to initiate migration, keratinocytes need to undergo significant alteration of their junctions and adhesion molecules; this includes detachment of hemidesmosomes and replacement of their collagen-binding receptors with new integrins that allow the keratinocytes to adhere to the newly formed provisional wound matrix. In addition, these keratinocytes begin to proliferate as they reduce production of proteins normally expressed by differentiated stratified epidermis, in particular specific keratins (see Ch. 56 ). During wound healing, one classic marker of activated keratinocytes is the expression of keratins associated with proliferation, keratins 6 (K6) and K16.
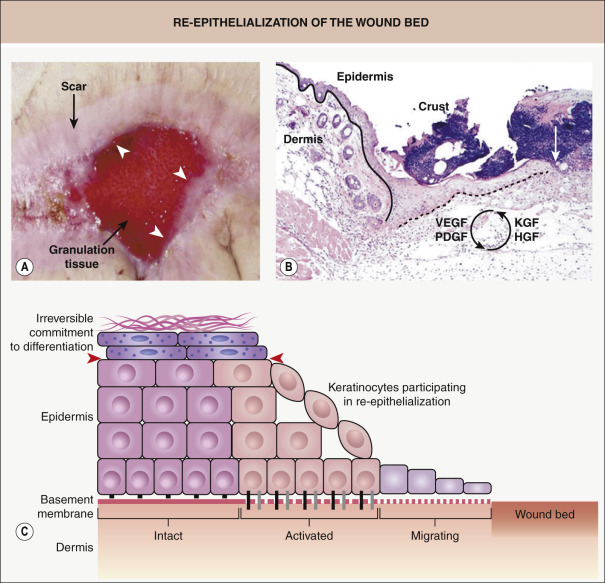
It has been proposed that the cytoskeletal changes associated with activation, including the expression of these particular keratin filaments, provide plasticity and flexibility, while still maintaining resilience of the intracellular scaffold that is important for migration . Many of the growth factors active during wound healing, such as keratinocyte growth factor (KGF) and hepatocyte growth factor (HGF), are potential regulators of these processes. These growth factors can be synthesized either by activated keratinocytes (and act in an autocrine loop) or by cells present in the dermis, in particular fibroblasts, and then act in a paracrine fashion .
As the stratified epithelium consists of keratinocytes committed to multiple stages of differentiation, it is important to know which keratinocytes can be activated for regeneration. Experimental data from mice expressing a transgene within defined keratinocyte subpopulations revealed that interfollicular epidermal stem cells, transient amplifying cells, and early differentiated cells have the ability to form a fully differentiated epidermis . These data suggest that primarily skin stem cells and transient amplifying cells contribute to wound healing and that keratinocytes become irreversibly committed to the differentiation process fairly rapidly and then respond poorly to activating signals. Once keratinocytes process profilaggrin into filaggrin, they undergo specific changes that mark late terminal differentiation, which seemingly demarcates the final “line of regenerative potency” within the differentiating keratinocyte population (see Fig. 141.4 ) .
More recent studies in mice which examined the contribution of stem cells to the formation of a new epidermis following injury concluded that there are multiple populations of stem cells residing within different locations of the epidermis and skin appendages. Under normal homeostatic conditions, stem cells residing in different locations maintain the differentiated lineages specific to those locations, i.e. hair follicle stem cells (HFSCs) maintain the hair lineages, sebaceous gland stem cells produce differentiated sebocytes, and stem cells within the interfollicular epidermis (IFE) give rise to the outermost barrier layers of the epidermis . Although in principle cells from all stem cell populations are capable of participating in epithelialization after a cutaneous injury, their specific contributions vary . The current view is that stem cells from the interfollicular epidermis as well as cells that lie within the lower bulge area (region of the hair follicle defined as the attachment site of the arrector pili muscle) and above the bulge (the isthmus) contribute long-lasting progeny during re-epithelialization, while cells located in the bulge play a minor role . These findings are supported by earlier studies in mutant mice in which epidermal wound repair still occurred despite the absence of hair follicles in mutant mice. Nonetheless, an acceleration of epidermal wound healing has been observed during the growth phase of the hair cycle. Collectively, these findings help to explain how depth of injury affects the capability of a wound to repair and to re-epithelialize.
Endothelial cells
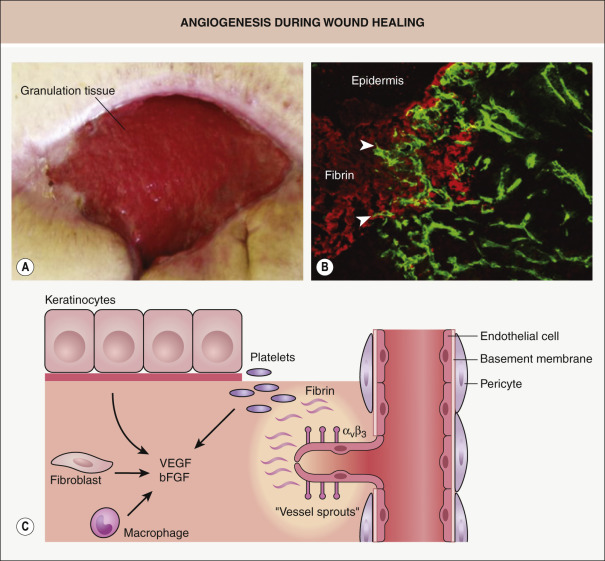
Activated and proliferating endothelial cells that form blood vessels normally express the α v β 3 integrin and make a significant contribution to angiogenesis, which is a vital component of normal wound healing ( Fig. 141.5 ) . New blood vessels ensure delivery of nutrients to meet the increased metabolic requirements of wound tissue and they help to reverse the hypoxic and acidotic milieu. Several features characterize neoangiogenesis within wounds: firstly, there is increasing evidence that this process occurs via both angiogenesis (sprouting of capillaries from existing blood vessels) and vasculogenesis (mobilization of bone marrow-derived endothelial progenitor cells; see Ch. 102 ). Secondly, the process is highly complex and requires a dynamic, temporally and spatially regulated interaction amongst endothelial cells, soluble angiogenic growth factors, and ECM molecules. Thirdly, wound angiogenesis is induced, at least in part, by the activation of the innate immune response. Immediately after injury, different subsets of leukocytes, in particular macrophages, are attracted to the wound site where they release multiple pro-angiogenic mediators (e.g. VEGF-A, IL-6, IL-8, TNF) that trigger and sustain the angiogenic response . Importantly, as shown for VEGF-A, the time of action, cellular source, and concentration of a given cytokine determine whether it activates, terminates, or inhibits angiogenesis. The delayed healing response observed in patients receiving anti-VEGF-A agents (e.g. bevacizumab) highlights the critical role VEGF-A plays in angiogenesis during tissue repair .