Basal Cell Nevus Syndrome: Introduction
|
Basal cell nevus syndrome (BCNS), also known as nevoid basal cell carcinoma syndrome and Gorlin syndrome [Online Mendelian Inheritance in Man (OMIM) #109400], is a rare autosomal dominant disorder associated with a panoply of phenotypic abnormalities that can be divided into developmental anomalies and postnatal tumors, especially basal cell carcinomas (BCCs).1 Although individual aspects had been reported previously, their syndromic association was first appreciated widely in the late 1950s.2,3
Epidemiology
The prevalence of BCNS is variously estimated to be 1 in 60,000 and 1 in 120,000 persons.4,5 The syndrome affects both sexes and occurs in a wide variety of cultural groups, and therefore does not have a predilection for a particular skin type. The condition appears to have complete penetrance but variable expressivity of traits, such that their clinical presentation within families is nonuniform. Further, as with many dominantly inherited conditions, new mutations are common. As a result, many patients may have no apparent affected ancestors or siblings.
Etiology and Pathogenesis
Almost all known BCNS patients thus far carry mutations in the PATCHED1 (PTCH1, UniGene Hs. 494538) gene residing on the long arm of chromosome 9.6,7 PTCH1 plays a central role in the hedgehog signaling pathway that is essential for the establishment of normal body and limb patterning in metazoan organisms.8 The PTCH1 locus behaves like a classic tumor suppressor gene (Fig. 116-1). The appearance of BCCs in small numbers at an older age in sporadic cases and in larger numbers at a younger age in patients with BCNS is reminiscent of differences in sporadic and hereditary cases of retinoblastoma.9 BCNS, like other tumor susceptibility syndromes, is inherited in an autosomal dominant manner, with inheritance of a loss-of-function allele followed by somatic loss of the remaining copy before tumor formation.
Figure 116-1
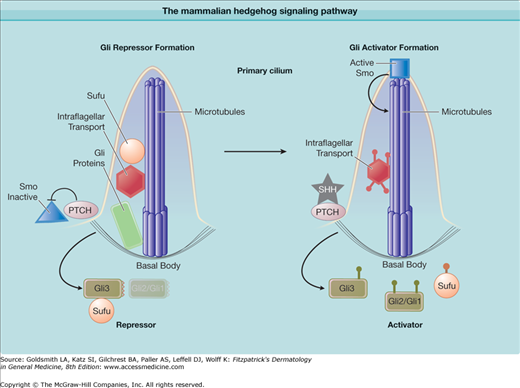
The mammalian hedgehog signaling pathway. PTCH1 is the receptor for the growth factor hedgehog and inhibits the function of smoothened (SMO) by sequestering it in an inactive state outside of the microtubule-based organelle, the primary cilum. Sonic hedgehog (SHH) inhibits PTCH1, allowing activation of SMO in the cilium. SMO inhibits Suppressor of Fused (SUFU), which in turn inhibits the GLI transcription factors GLI1, GLI2, GLI3. Intraflagellar transport proteins help mediate GLI processing.
Identification of the gene mutation responsible for BCNS facilitated a molecular verification of the predicted PTCH1 tumor suppressor function.6,7 BCNS patients inherit one defective PTCH1 allele and their tumors contain an additional somatic mutation. As with other tumor suppressor genes, PTCH1 mutations have also been found in older adults with sporadic BCCs and other sporadic tumors known to be overrepresented in BCNS patients (e.g., medulloblastomas and meningiomas), which supports the idea that two somatic “hits” are required in sporadic tumors.
In contrast, the two-hit genetic model proposed for PTCH1-dependent tumor development may not fully explain the developmental abnormalities seen in BCNS, which occur earlier. PTCH1 heterozygote mice display many of the developmental abnormalities seen in BCNS patients despite a wild-type copy of the nonaffected allele.10,11 This argues that certain tissues (skeleton, limb, neural tube) require higher levels of PTCH1 to regulate tissue development and that heterozygotes cannot compensate for loss of the other allele (haploinsufficiency). In addition, the PTCH1 locus appears to be regulated in part by silencing. Recent observations indicate that methylation blockers can reactivate PTCH1 expression shut off during tumor formation, suggesting that silencing of PTCH1 expression may also play a role in normal development.12
Are PTCH1 mutations the only cause of BCNS? Despite direct sequencing of PTCH1 exons in patients, somatic mutations are found in fewer than half of BCCs and germ-line mutations in fewer than 100% of BCNS patients.13,14 This suggests that mutations in other unidentified genes may underlie the disease. However, early linkage studies found a strong association with the PTCH1 locus. Moreover, although mutations are not found in PTCH1 exons in some patients, hedgehog signaling pathway abnormalities are still present in these tumors. Recently, one family with BCNS and atypical features (medulloblastoma, plantar pits, but no BCCs) was found to have germ-line mutations in Suppressor of Fused (SUFU)—a tumor suppressor gene and negative regulator of sonic hedgehog (SHH) signaling (Fig. 116-1).15 This suggests mutations may be found in other members of the signaling pathway in rare cases.
PTCH1 protein plays a critical role in the hedgehog-signaling pathway (Fig. 116-1). Genetic and biochemical studies in Drosophila and mammals indicate that PTCH1 protein inhibits this signaling pathway by inhibiting the function of the central G protein-coupled receptor smoothened (SMO), and that the extracellular ligand hedgehog abrogates this inhibition. Signaling by SMO results in the activation of the Glioma associated oncogene (GLI) family of zinc finger proteins that mediate all the transcriptional effects of hedgehog signaling. Three GLI proteins mediate activation and suppression of hedgehog target genes, with GLI1 and GLI2 acting as activators and GLI2 and GLI3 as suppressors. SMO signaling tips the balance toward activation and away from target gene suppression. In mammals, GLI activity is controlled by the novel cytoplasmic protein SUFU, which promotes the transcriptional repression and inhibits activation.16 SMO inhibits SUFU activity, thus releasing GLI proteins to become transcriptional activators. When hedgehog binds to PTCH1, PTCH1 inhibition of SMO is relieved and the pathway is activated. Thus, loss of PTCH1 function allows unregulated SMO activity and initiates tumor formation.
How PTCH1 functions as a tumor suppressor is still under investigation. A significant function of PTCH1 is to inhibit SMO, although how PTCH1 accomplishes this is yet unknown. PTCH1 acts in an enzymatic manner as only a few molecules are necessary to inhibit SMO by several fold.17 Moreover, PTCH1 and SMO are thought to exist in distinct endosomes and thought not to physically interact. Recent studies demonstrate that components of the hedgehog pathway are localized to a novel microtubule based organelle called the primary cilium (Fig. 116-1).18 Mutations that affect the structure and function of the cilium, known collectively as the human ciliopathies, are also known to disrupt maximal hedgehog signaling and the patients have some similarities to BCNS. SMO accumulates in the cilia via intraflagellar transport proteins and this accumulation is correlated with pathway activation. PTCH1 is localized at the base of the organelle and appears to prevent SMO entry. Loss of PTCH1 or mutations that allow SMO to move into the cilium result in increased pathway activity.19 PTCH1 shares homology with sterol-sensing domain-containing proteins such as SREBP (sterol regulatory element-binding protein) and β-hydroxy-β-methylglutaryl–coenzyme A reductase.20 It also shares significant structural identity with the resistance-nodulation-division (RND) permease family of small-molecule transporters, which suggests that PTCH1 may act as a molecular pump. This evokes the tantalizing hypothesis that PTCH1 regulates SMO entry and activation within the cilium by regulating the production or distribution of small-molecule second messengers in the cell.
The identification of mutations in members of the SHH pathway in both BCNS patients and in sporadic BCCs supports the central role of SHH target gene induction in BCC pathogenesis. PTCH1 mutations are by far the most common and are found in approximately 40% of tumors.21 Epidemiologic evidence implicates ultraviolet light in the pathogenesis of sporadic BCCs. UV signature mutations have been found in PTCH1 and these provide additional evidence for a role of sunlight in cancer development.22
Mutations of SMO protein have been identified in approximately 10% of BCCs, and these mutations appear to render SMO protein resistant to PTCH1 inhibition.23 Indeed, experimental transfection of cells with mutant SMO sequences can transform them to a malignant phenotype. This finding that BCCs may have upregulation of hedgehog target gene expression due to mutations in either PTCH1 or SMO argues that it is the upregulation of hedgehog signaling rather than the specific mutation that is crucial to BCC formation. Consistent with this is the finding that mutations in the gene encoding SUFU have been reported to underlie formation of medulloblastomas and BCNS.15,24
The identification of these molecular abnormalities in BCCs for the first time has permitted the development of mouse models of this tumor. Previously, tumor-initiating insults to mouse skin (ultraviolet, ionizing radiation or chemical carcinogens) have produced papillomas and carcinomas of the squamous, but not the basal cell lineage. Mouse models that show spontaneous development of BCC-like tumors include those with epidermal overexpression of hedgehog,25 of mutant SMO,23 or of GLI126,27 or GLI2.28,29 In addition, ptc1+/− mice, which, like BCNS patients, have one instead of two functioning alleles of PTCH1, not only develop BCCs and related tumors but also develop plantar pits, medulloblastomas, and rhabdomyosarcomas, as do BCNS patients. The mouse BCCs (mimicking human BCCs) occur spontaneously in low numbers and in small sizes, but in much higher numbers and of larger size in mice exposed to ultraviolet or ionizing radiation.30
Clinical Findings
Hedgehog signaling plays a critical role in the expansion of progenitor cells in a wide variety of tissues in both invertebrate and vertebrate organisms. PTCH1 normally is expressed both during development and in the adult, which suggests an ongoing postnatal role. Studies involving experimental models inappropriately expressing components of the hedgehog signaling pathway have revealed significant developmental anomalies.31 The resemblance of these anomalies to those characteristic of BCNS implies that aberrant activation of the hedgehog signaling pathway is a sufficient explanation for the developmental and tumorigenic anomalies of BCNS, even if the precise pathogenic mechanisms have yet to be elucidated.
Patients with BCNS show multiple abnormalities, none of which is unique to this syndrome (eTable 116-0.1).5,13,14 The three abnormalities considered to be most characteristic of the syndrome are tumors such as medulloblastomas or BCCs, pits of the palms and soles, and odontogenic cysts of the jaw.
IGR14 (France) | Evans et al36 (United Kingdom) | Shanley et al5 (Australia) | Kimonis et al13 (USA) | |
|---|---|---|---|---|
Number of cases | 22 | 84 | 118 | 105 |
Number of families | 5 | 29 | 64 | 26 |
Average age (year) | 44.9 | 35 | 34.5 | |
Sex ratio (Male : Female) | 1:1.75 | 1:1.3 | 1:1.3 | 1:1.2 |
BCC (%) | 100 | 47 | 76 | 80 |
Average age at first BCC (year) | 24.2 | 20.3 | 21.4 | |
Palmar pits (%) | 45 | 71 | 80 | 8 |
Dental cysts (%) | 62 | 66 | 75 | 74 |
Falx calcification (%) | 66 | 92 | 65 | |
Epidermal cysts (%) | 41 | 51 | ||
Craniofacial Anomalies | ||||
Macrocephaly (%) Skull protrusion (%) Cleft palate (%) | 27 18 | 5 | 80 66 4 | 49 26 3 |
Ophthalmologic Anomalies | ||||
Hypertelorism (%) Strabismus (%) | 18 4.5 | 6 | 42 19 | |
Skeletal Anomalies | ||||
Pectus excavatum (%) Scoliosis (%) Kyphosis (%) Cervical ribs (%) Bifid ribs (%) Spina bifida (%) Vertebral fusion (%) | 23 18 9 5 16 9 4.5 | 23 | 12 31
16 26 19 10 | |
Neurologic Anomalies | ||||
Anosmia (%) Deafness (%) Corpus callosum tumor (%) | 4.5 4.5 9 | 9 |
10 | |
Tumors | ||||
Medulloblastoma (%) Meningioma (%) Ovarian fibroma (%) | 13 18 13 | 4 1 24 | 1
14 | 4 5 17 |
Individual BCCs from patients with BCNS cannot be distinguished from those in sporadic cases (see Chapter 115), which is not surprising in view of the similar pathogenesis in familial and sporadic cases. What is distinguishing is their appearance in large numbers starting at an early age. They may be banal appearing and confused grossly with nevocytic nevi—hence the name basal cell nevus. They may also have a translucent, papulonodular appearance more characteristic of sporadic BCCs and may invade locally. In rare cases, they may even metastasize and cause death. Although the ratio of sun-protected to sun-exposed BCCs may be higher in BCNS than in sporadic cases, sunlight and ionizing radiation clearly accelerate BCC formation in BCNS patients, and darkly pigmented BCNS patients may have few to no BCCs (Figs. 116-2, 116-3, and 116-4).
Figure 116-2








