Most of the clinical and microscopic characteristics of congenital and hereditary hair shaft abnormalities were well described many years ago. Because of this and because many of these conditions have obvious pedigrees, the targeted topical selective gene therapy being developed for hair follicle faults is feasible. Impressive progress has been made in our knowledge of genes that control normal development and anomalies and the search is on to define these genes in hair diseases.
It is not the purpose of this chapter to cover all hypotrichotic syndromes. We cover most of those that have been classified and reviewed the literature to select others that require their undescribed aspects to be explored further.
Congenital Atrichia and Hypotrichosis
Atrichia congenita is characterized by follicular agenesis or programmed follicular destruction. A child may be born with complete absence of scalp and body hair or may progress to that stage in the first 5 years of life. In another variant of the disease, a neonate is born with lanugo hair, which is shed in the first few months of life and never replaced. Caution should be exercised to ensure that a hair abnormality is isolated because other associations may be unveiled only over time.
Congenital hypotrichosis is a less severe form of atrichia congenital; hair is diffusely thinned but not absent. This form usually occurs as an isolated defect. Hypotrichosis may not be noticed until the age of 2 years because of variations in the quality and quantity of hair normally present at birth.
Most cases of congenital hypotrichosis and atrichia congenita are autosomal recessive (AR) traits. Several autosomal dominant (AD) pedigrees have been identified. When no family history is obtained, a scalp biopsy should be performed to exclude alopecia areata totalis. A biopsy will also provide information about follicle architecture and count and reveals other cutaneous abnormalities.
The conditions that present with hypotrichosis without complete alopecia in infancy constitute a very long list. The hypotrichosis may be secondary to follicular hypoplasia or to faulty hair shaft production and breakage. Many of the ectodermal dysplasias are associated with hypotrichosis, but unfortunately most hair shaft abnormalities have not been well characterized. Abnormal hair is generally described clinically only as brittle, sparse, or lusterless.
Numerous attempts have been made to classify the conditions characterized by congenital alopecia or hypotrichosis. In 1892, Bonnet [1] proposed the first known classification based on embryological principles. It has been widely used until now and roughly divides congenital hypotrichoses with normal ectodermal structures from those with associated teeth and nail defects. Cockayne [2] and Muller [3] later attempted a more critical analysis by proposing a working classification that allowed the currently named syndromes to be identified and provided provisional status for those not yet characterized. After the Berlin Congress in 1981, Sâlamon [4] proposed a classification for the global problem of hair loss that is considered one of the most useful systems for the study of congenital hypotrichosis.
In recent years, major breakthroughs in genomic techniques yielded considerable progress toward unraveling the molecular bases of inherited skin diseases. At present, over 6000 Mendelian disorders are known; 560 involve skin abnormalities and are associated with more than 500 unique protein-coding genes. More than 300 inherited disorders featuring hair abnormalities have been catalogued to date, and yet no genetic defects have been identified in a substantial number of the disorders (Figures 2.1 and 2.2) [5].
FIGURE 2.1 Many congenital hypotrichoses not associated with other defects cannot be classified properly.
This new knowledge led to a reorganization of the classification systems for this group of diseases. Instead of being based solely on clinical findings, the classification now integrates both clinical and molecular features. It is now possible to assign most forms of hypotrichosis to one gene (or group of genes) on the basis of limited information about three clinical features: mode of inheritance, presence or absence of associated features, and microscopic appearance of hair shafts. When such data are available, it is easy to decide the optimal molecular diagnostic strategy and order genetic testing that is likely to lead to a correct diagnosis (Table 2.1). As Betz et al. [5] indicate, most patients with hypotrichoses can be assigned to a group of molecularly defined hair disorders using three clinical criteria.
Clinicogenetic Classification of Hypotrichoses
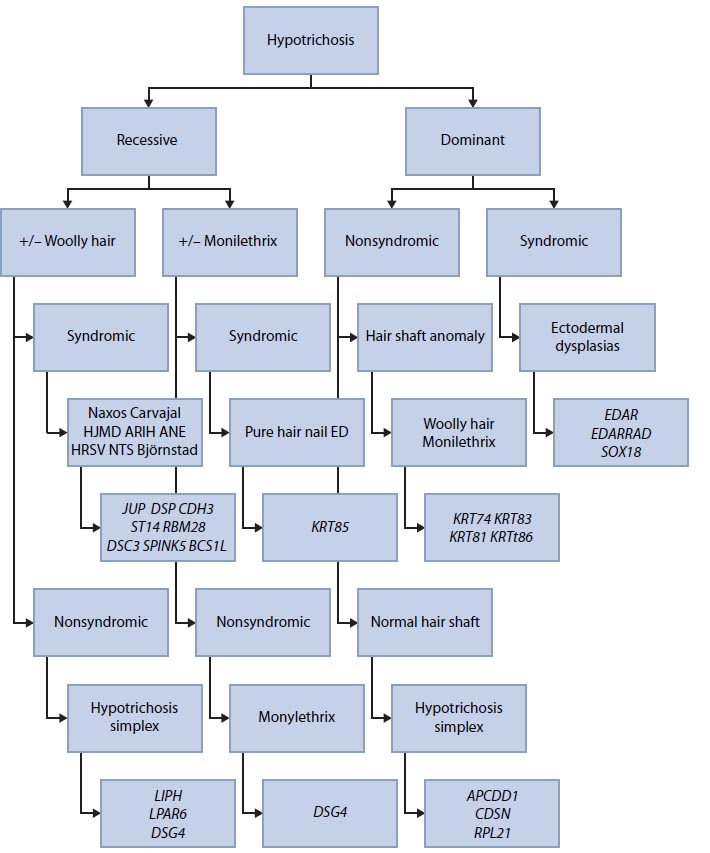
Source: Adapted from Betz RC et al. J Invest Dermatol 132: 906–914, 2012.
Note: Using three clinical criteria, most patients with hypotrichosis can be assigned to a group of molecularly defined hair disorders. ANE: Alopecia, neurological defects, and endocrinopathy; ARIH: Autosomal recessive ichthyosis with hypotrichosis; DSG4: Desmoglein 4; ED: Ectodermal dysplasia; EDAR: Ectodysplasin A receptor; EDARADD: EDAR-associated death domain; HJMD: Hypotrichosis with juvenile macular dystrophy; HRSV: Hypotrichosis with recurrent vesicles; KRT85: Keratin 85; LIPH: Lipase H; LPAR6: Lysophosphatidic acid receptor 6; NTS: Netherton syndrome.
FIGURE 2.2 In non-classified congenital hypotrichosis, no hair shaft alterations are present.
In this review, we will follow the practical classification based on the clinical observations proposed by Camacho [6]. Readers should be aware, however, that each group covers a large clinical spectrum of disorders that are not grouped on a pathogenetic basis. The classification scheme in Table 2.2 is largely of didactic value.
Classification of Generalized Congenital and Hereditary Alopecias
1. | Genodermatoses with non-scarring hypotrichosis | ||
1.1 | With skeletal alterations | ||
McKusich disease or chondrodysplasia | |||
Moynahan disease (hypotrichosis, syndactyly, retinitis) | |||
Trichorhinophalangeal syndromes | |||
Pierre-Robin syndrome | |||
Cardio-facial cutaneous syndrome | |||
Alopecia-contracture-dwarfism (ACD) syndrome with mental retardation | |||
Oculo-dental-digital syndrome | |||
Dubowitz syndrome | |||
Noonan syndrome | |||
Hallemann-Streiff syndrome | |||
1.2 | With ectodermic alterations | ||
Ectodermal dysplasias | |||
1.3 | With neuroectodermal alterations | ||
Tricothiodystrophy | |||
1.4 | With chromosomal alterations | ||
Down syndrome | |||
Klinefelter syndrome | |||
Turner syndrome | |||
1.5 | With amino acid metabolism alterations | ||
Hypotrichosis, hair shaft defect, hypercysteine hair, and glucosuria syndrome | |||
Citrulinemia | |||
Hartnup disease | |||
Homocystinuria | |||
Phenylketonuria | |||
Tyrosinemias I and II | |||
1.6 | Other genodermatoses with hypotrichosis | ||
1.6.1 | Progerias | ||
Werner syndrome or pangeria | |||
Hutchinson-Gilford syndrome or childhood progeria | |||
Variot-Cailleau syndrome or childhood gerodermia | |||
Other progerias | |||
1.6.2 | Others | ||
Congenital ichthyosiform erythrodermia | |||
Netherton syndrome | |||
Tay syndrome | |||
Rud syndrome | |||
KID syndrome | |||
Rothmund-Thomson disease | |||
Poikilodermia–alopecia–retrognathism–cleft palate syndrome | |||
Zinsser-Cole-Engman disease | |||
Kallin syndrome or epidermolysis bullosa simplex | |||
1.7 | Genodermatoses with hypotrichosis and tumors | ||
Rombo syndrome | |||
Bazex-Dupré-Christol syndrome | |||
1.8 | Hereditary simple hypotrichosis | ||
2 | Genodermatoses with scarring hypotrichosis | ||
2.1 | Darier disease | ||
2.2 | Ichthyosis X | ||
2.3 | Dystrophic epidermolysis bullosa | ||
2.4 | Incontinentia pigmenti | ||
2.5 | Polyostotic fibrous dysplasia | ||
2.6 | Conradi syndrome | ||
2.7 | Happle syndrome | ||
Generalized Congenital Alopecia
Genodermatoses with Non-Scarring Hypotrichosis
Genodermatoses with Skeletal Alterations
Trichorhinophalangeal Syndromes
Trichorhinophalangeal syndromes (TRPSs) constitute distinctive combinations of hair, facial, and bone abnormalities with AD inheritance. Three types are described below.
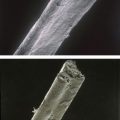
Trichorhinophalangeal syndrome type I is characterized clinically by a variable congenital hypotrichosis, piriform (pear-shaped) nose, coniform epiphysis, subnasal fold, thin lips, prognatia, and mandibular hypoplasia (Figure 2.3). The hair alterations consist of diffuse alopecia with a broad forehead and partial alopecia of the lateral third of the eyebrows. Scanning electron microscopic studies of hair shafts can reveal flattened hairs with an elliptoid transverse section pattern. Mechanical behavior of the hair may be abnormal and reveal a significant increase in the viscous parameter indicating decreased intermolecular bridging within the keratin matrix [7].
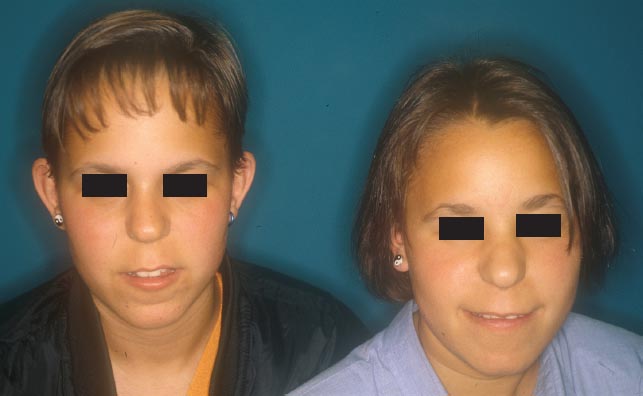
FIGURE 2.3 In trichorhinophalangeal syndrome, the face displays characteristic dwarfism, diffuse hypotrichosis, piriform nose, and elongated philtrum.
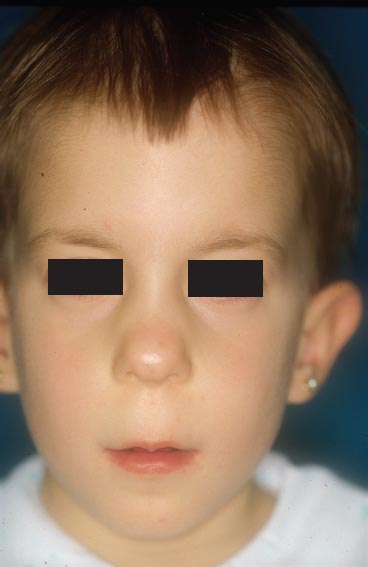
FIGURE 2.4 Typical facies of trichorhinophalangeal syndrome with piriform nose.
Trichorhinophalangeal syndrome type II (Langer-Giedion syndrome). Patients with type II usually present with hypotrichosis of the scalp hair, piriform nose, and redundant skin as in type I along with multiple cartilaginous exostoses. Lu et al. [8] described alterations to this syndrome including aplasia of the epiglottis and congenital nephrotic syndrome.
Trichorhinophalangeal syndrome type III is a newly defined clinical entity [9] inherited as an AD trait and clinically characterized by growth retardation, craniofacial abnormalities, severe brachydactyly, and sparse hair. The absence of mental retardation and cartilaginous exostoses is required for the diagnosis of type III. Other associated abnormalities include short stature, a thin upper lip and prominent lower lip, a pear-shaped nose, stubby fingers and toes with cone-shaped epiphyses, and sparse scalp hair (Figures 2.4 through 2.6).
Dubowitz Syndrome
First described in 1965, Dubowitz syndrome (DS) is characterized by a peculiar face, eczema, small stature, and mild microcephaly. The cutaneous findings consist of eczematous eruptions affecting the face and flexural areas [10]. Scalp hair is sparse and brittle and commonly affects the lateral eyebrows. Patients affected by DS have moderate mental deficiencies with a tendency toward hyperactivity, short attention spans, stubbornness, and shyness and have been characterized by their high-pitched weak cries.
Hallermann-Streiff Syndrome
Hallermann-Streiff syndrome is a rare congenital anomaly characterized by a peculiar bird face, mandibular and maxillary hypoplasias, dyscephaly, congenital cataracts, microphthalmia, hypotrichosis, skin atrophy, and short stature [11]. Dental abnormalities are present in 80% of cases and include malocclusion, crowding, severe caries, supernumerary and neonatal teeth, enamel hypoplasia, hypodontia, premature eruption of primary dentition, agenesis of permanent teeth, and anterior displacement or absence of condyles [12].
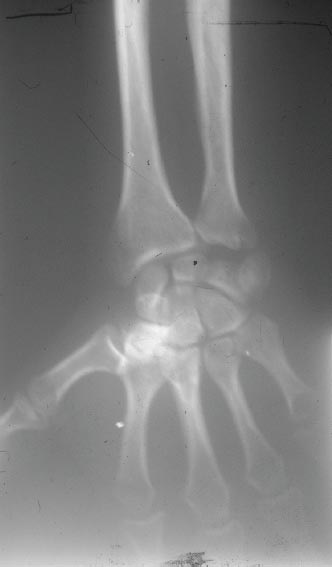
FIGURE 2.5 Radiography reveals cone epiphysis of first phalanges and shortening of some metatarsi.
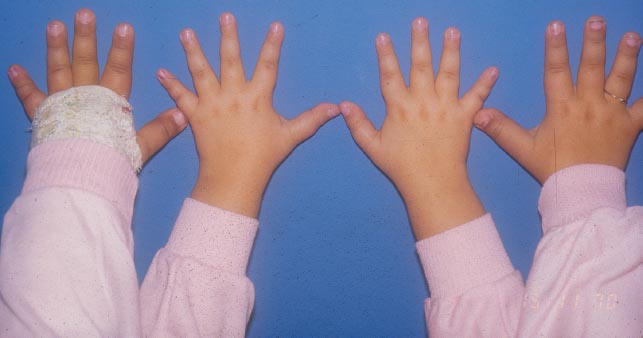
FIGURE 2.6 Clinical observation of hands showing shortening of metatarsi.
Genodermatoses with Ectodermal Alterations
Ectodermal Dysplasias
Ectodermal dysplasias (EDs) are a heterogeneous group of conditions primarily affecting the hair, teeth, nails, and skin, and are classified according to the tissues affected. EDs are rare diseases with an estimated incidence of only 7 in 10,000 births [13]. Of the 170 EDs described to date, fewer than 30 have been explained at the molecular level via identification of the causative genes.
The ectodermal dysplasia term was originally applied to anhidrotic ectodermal dysplasia in which hair, teeth, nails, and sweat glands are defective. The classification proposed by Freire-Maia in 1977 [14] was based on a primary defect of ectodermal derivatives. Conditions in which the ectodermal changes are secondary as in xeroderma pigmentosum are thus excluded from the EDs. According to Freire-Maia’s classifications, subgroup 1 is hair dysplasia; subgroup 2, dental dysplasia; subgroup 3, nail dysplasia; subgroup 4, sweat gland defects; and subgroup 5, defects of other ectodermal structures. In 1980, Solomon and Keuer [15] defined ED subgroups based on the ectodermal structures affected (Table 2.3).
Anhidrotic Ectodermal Dysplasia (Christ-Touraine Syndrome)
In this X-linked syndrome, sweat glands and other ectodermal-derived appendages are absent or few in number. The full syndrome occurs only in males. Scalp and body hair is short, fine, and very sparse and often brightly colored; it may increase in quantity after puberty. Eyebrows and eyelashes may also be sparse or absent but may be minimally affected. The prominent square forehead, saddle nose, thick lower lip, and pointed chin produce a distinctive face. The skin around the eyes is finely wrinkled and may be pigmented. The teeth may be absent or few in number, and the canines and incisors are characteristically cone shaped.
Ectodermal Dysplasia Subgroups Proposed by Solomon and Keuer
Subgroups 1, 2, 3, and 4 (hair, teeth, nails, and sweating defects) Anhidrotic ectodermal dysplasia Rapp-Hodgkin Ectrodactyly–ectrodermal dysplasia–cleft palate syndrome Popliteal web syndrome Xeroderma–talipes–enamel defect syndrome |
Subgroups 1, 2, and 3 (hair, teeth, and nail defects) Clouston dysplasia Trichodento-osseous syndrome Ellis-van Creveld syndrome Ankyloblepharon–ectodermal defect–cleft palate syndrome Basan syndrome Tooth–nail syndrome |
Subgroups 1, 3, and 4 (hair, nails, and sweating defects) Freire-Maia syndrome |
Subgroups 1 and 2 (hair and teeth defects) Orofaciodigital syndrome I Sensenbrenner syndrome Trichodental syndrome |
Subgroups 1 and 3 (hair and nail defects) Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|