Other T/NK-cell lymphomas that may involve the skin994
Other B-cell lymphomas that may involve the skin996
Precursor B-lymphoblastic leukemia/lymphoma996
Chronic lymphocytic leukemia/small lymphocytic lymphoma (B-CLL)996
Mantle cell lymphoma997
Primary effusion lymphoma997
Lymphoplasmacytic lymphoma/Waldenström’s macroglobulinemia (LL/M)997
Burkitt and Burkitt-like lymphoma998
Plasmacytoma and secondary myeloma998
Lymphoid hyperplasias mimicking primary lymphoma1000
Cutaneous lymphoid hyperplasia simulating B-cell lymphoma1000
Lymphomatoid drug reactions1001
Reactions resembling CD30+ lymphoproliferative disorders1002
Pseudolymphomatous folliculitis1002
Jessner’s lymphocytic infiltrate1002
Acral pseudolymphomatous angiokeratoma1003
Cutaneous CD8+ T-cell infiltrates in HIV/AIDS1003
Miscellaneous1003
INTRODUCTION
The diagnosis and classification of cutaneous lymphomas remain one of the most challenging areas of dermatopathology. At the time the last edition of this book was published, the WHO classification of tumors of hematopoietic and lymphoid tissues based on the REAL classification of lymphomas was being published.1. and 2. This classification included some cutaneous lymphomas as subgroups within the main classification framework. At the same time, the Project Group of the European Organization for Research and Treatment of Cancer published a classification of cutaneous lymphomas which came to be known as the EORTC classification of cutaneous lymphomas. 3 Those two classifications were based on a combination of histological, clinical, immunohistological, and cytogenetic features. There were certain differences in terminology and emphasis between these two classifications and the appropriateness of one or other of the classifications was debated in the literature.4.5.6.7.8.9.10.11. and 12. The EORTC classification included only those lymphomas which appeared to be unique to the skin. The REAL/WHO classification had the advantage of including other lymphomas which involved the skin secondarily or rarely as ‘primary’ cutaneous lymphomas. 13 Other proposals were made for a stand-alone classification of cutaneous lymphomas based on the REAL/WHO classification.13. and 14.
Recently, a consensus classification of cutaneous lymphomas, the WHO/EORTC classification of cutaneous lymphomas, has been published15. and 16. and critiqued in the literature.16.17. and 18. This compromise classification defines ‘primary cutaneous lymphomas’ as lymphomas that present in skin with no evidence of extracutaneous disease at the time of diagnosis. There are some inherent problems with this strict definition of primary cutaneous lymphoma as some putative primary cutaneous lymphomas are associated with systemic spread to other systems at the time of presentation, e.g. Sézary syndrome and adult T-cell leukemia/lymphoma. The definition does not encompass secondary cutaneous lymphomas. Nevertheless this classification is widely used and will be used as the framework for the discussion in this chapter (see Table 41.1). Other lymphoma entities which only rarely present in the skin or do so as part of systemic spread of lymphoma from another site are classified according to the general WHO classification of lymphomas.
| Cutaneous T-cell and NK-cell lymphomas |
Mycosis fungoides and subtypes Folliculotropic mycosis fungoides Pagetoid reticulosis Granulomatous slack skin Hydroa vacciniforme-like lymphoma Sézary syndrome Adult T-cell leukemia/lymphoma Primary cutaneous CD30+ lymphoproliferative disorders Primary cutaneous anaplastic large cell lymphoma Lymphomatoid papulosis Subcutaneous panniculitis-like T-cell lymphoma Extranodal NK/T-cell lymphoma, nasal type Primary cutaneous peripheral T-cell lymphoma, unspecified Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma (provisional) Cutaneous γ/δ T-cell lymphoma (provisional) Primary cutaneous CD4+ small/medium pleomorphic T-cell lymphoma (provisional) |
| Cutaneous B-cell lymphomas |
Primary cutaneous marginal zone B-cell lymphoma Primary cutaneous follicle center cell lymphoma Primary cutaneous diffuse large B-cell lymphoma, leg type Primary cutaneous diffuse large B-cell lymphoma, other Intravascular large B-cell lymphoma Plasmablastic lymphoma T-cell/histiocyte-rich B-cell lymphoma Lymphomatoid granulomatosis |
| Precursor hematologic neoplasm |
| Blastic plasmacytoid dendritic cell neoplasm |
| Other T/NK-cell lymphomas that may involve the skin |
Precursor T-lymphoblastic lymphoma/leukemia T-cell prolymphocytic leukemia Angioimmunoblastic T-cell lymphoma Primary systemic anaplastic large cell lymphoma Intravascular T- and NK-cell lymphoma Aggressive NK-cell leukemia Other T/NK-cell lymphomas and leukemias |
| Other B-cell lymphomas that may involve the skin |
Precursor B-lymphoblastic leukemia/lymphoma Chronic lymphocytic leukemia/small lymphocytic lymphoma Mantle cell lymphoma Primary effusion lymphoma Lymphoplasmacytic lymphoma/Waldenström’s macroglobulinemia Burkitt and Burkitt-like lymphoma Plasmacytoma and secondary myeloma |
| Other lymphomas |
| Hodgkin lymphoma |
| Cutaneous infiltrates of leukemias |
| Myeloid leukemias, myeloproliferative diseases and myelodysplastic syndromes |
| Lymphoid hyperplasia mimicking primary lymphoma |
Lymphoid hyperplasia simulating B-cell lymphoma Lymphomatoid drug reactions Reactions resembling CD30+ lymphoproliferative disorders Pseudolymphomatous folliculitis Jessner’s lymphocytic infiltrate Acral pseudolymphomatous angiokeratoma Cutaneous CD8+ T-cell infiltrates in HIV/AIDS |
| Cutaneous infiltrates in post-transplant lymphoproliferative disorders |
| Other lymphoproliferative disorders associated with immunosuppression |
Methotrexate-associated lymphoproliferative disorders Cutaneous infiltrates in HIV/AIDS |
| Miscellaneous |
| Extramedullary hematopoiesis |
The current diagnosis of lymphomas requires some knowledge of lymphocyte ontogeny and techniques which are used to demonstrate such aspects as cell phenotype, clonality of a proliferation, and cytogenetic features including the presence of viral genetic material. Most laboratories are now able to perform these techniques, including immunohistochemistry, in-situ hybridization, polymerase chain reaction technology (PCR), and fluorescence in-situ hybridization (FISH).19.20.21. and 22. Flow cytometry is also used for immunophenotyping23 but unlike other organs, the number of neoplastic cells present in skin is sometimes small and cells are difficult to extract, particularly those in the epidermis. Enzymatic and mechanical disintegration methods have been used to extract cells. 24
Other techniques, such as gene expression profiling using cDNA microarray technology and serum proteomics, are being used to more clearly define subgroups of cutaneous lymphoma, distinguish between cutaneous lymphoma and inflammatory dermatoses, and identify prognostic groups.25.26.27.28.29. and 30.
The significance of established clonality of either B or T cells in cutaneous lymphoid infiltrates remains a problem of interpretation.31. and 32. Although it has often been stated that lymphocyte clonality is not equivalent to malignancy33 demonstration of clonality in the appropriate cellular infiltrate and clinical scenario should be regarded as lymphoma.3. and 34. Follow-up studies on atypical lymphoid infiltrates with demonstrated monoclonal populations of T or B lymphocytes have shown progression to clear-cut lymphoma in some cases.34. and 35. Follow-up may be needed over a long period because of the slow evolution and indolent behavior of many cutaneous lymphomas. Patients with a primary cutaneous lymphoma have an increased risk of developing another lymphoproliferative disorder. 36
Most antibodies used for phenotyping and assessment of proliferation are available for use on paraffin-embedded tissue. Currently used antibodies and their various specificities are listed in Table 41.2. 37
| Antibody | Predominant cells |
|---|---|
| CD1a | Langerhans cells, Langerhans cell histiocytosis, some T-lymphoblastic lymphomas |
| CD2 | T cells and T-cell lymphomas |
| CD3 | T cells and T-cell lymphomas |
| CD3ε (cytoplasmic CD3) | T cells, NK cells, T-cell neoplasms, NK-cell neoplasms |
| CD4 | T-helper cells, monocytes, macrophages, Langerhans cells, peripheral T-cell lymphomas, MF, HTLV-1 associated adult T-cell leukemia/lymphoma |
| CD5 | T cells, T-cell lymphoma, B-cell chronic lymphocytic leukemia/small lymphocytic lymphoma, mantle cell lymphoma |
| CD7 | T cells, T-cell lymphomas, myeloid leukemia, NK-cell neoplasms, T-cell lymphoblastic lymphoma/leukemia |
| CD8 | T-cytotoxic/suppressor cells, NK cells, T-cell lymphomas, e.g. subcutaneous panniculitis-like T-cell lymphoma |
| CD10/(CALLA) | Precursor B cells, B-lymphoblastic leukemia/lymphoma, follicle center cell/follicular lymphoma |
| CD15 | Neutrophils, monocytes, Reed–Sternberg cells (classical Hodgkin lymphoma), acute myeloid leukemia |
| CD20 | B cells, B-cell lymphomas |
| CD21 | Follicular dendritic cells, follicular dendritic cell neoplasms, mantle and marginal zone B cells |
| CD23 | Follicular dendritic cells, mantle zone B cells, B-small lymphocytic lymphoma/chronic lymphocytic leukemia |
| CD30 | Activated lymphoid cells, anaplastic large cell lymphoma, lymphomatoid papulosis, Reed–Sternberg cells (classical Hodgkin lymphoma), embryonal carcinoma |
| CD43 | T cells, myeloid cells, mast cells, T-cell lymphomas, some B-cell lymphomas, myeloid leukemia, mast cell neoplasms |
| CD45 (leukocyte common antigen, LCA) | Hematolymphoid cells, most B- and T-cell lymphomas |
| CD45RO | T cells, histiocytes, myeloid cells, T-cell lymphomas, histiocytic neoplasms, myeloid leukemias |
| CD56 | NK cells, subset of activated T cells, T/NK-cell neoplasms, subset of T-cell lymphoma, cutaneous γδ T-cell lymphoma |
| CD57 | NK cells, T-cell subset, subset of T-cell neoplasms, NK-cell neoplasms |
| CD68 | Histiocytes, myeloid cells, mast cells and their neoplasms |
| CD79a | Immature and mature B cells, B-cell lymphomas, plasma cells, plasma cell neoplasms |
| CD99 | T- and B-lymphoblastic lymphoma/leukemia, small round cell tumors (Ewing’s sarcoma/PNET, synovial sarcoma, and others) |
| CD117 (KIT gene product) | Mast cells and mast cell disorders, some myeloid leukemias, Merkel cell carcinoma |
| CD123 | Plasmacytoid dendritic cells, blastic plasmacytoid dendritic cell neoplasm |
| CD138 | Plasma cells, some B immunoblasts, plasma cell neoplasms, some carcinomas |
| ALK-1 | Some types of anaplastic large cell lymphoma |
| βF1 | Major population of T cells (αβ T cells) not NK cells, most T-cell neoplasms |
| Bcl-2 | Non-germinal center B cells, most T cells, most follicular lymphomas but only some cutaneous follicle center cell lymphomas, many other B-cell neoplasms |
| Bcl-6 | Germinal center B cells, follicular lymphoma including cutaneous follicle center cell lymphoma |
| Bob.1 | B cells including plasma cells, B-cell neoplasms, plasma cell neoplasms |
| Cyclin D1 | Some histiocytes, mantle cell lymphoma |
| Epithelial membrane antigen (EMA) | Plasma cells, plasma cell neoplasms, anaplastic large cell lymphoma, lymphomatoid papulosis, nodular lymphocyte predominant Hodgkin lymphoma, many epithelial neoplasms, epithelioid sarcoma |
| EBV-latent membrane protein-1 (LMP-1) | Hodgkin lymphoma, post-transplant lymphoproliferative disorders, some NK/T-cell neoplasms |
| Granzyme B, perforin, TIA-1 | NK cells, cytotoxic T cells, several T-cell and NK-cell neoplasms |
| Immunoglobulin light chains (kappa and lambda) | Plasma cells, plasma cell neoplasms, plasmacytoid neoplasms |
| Ki-67 | Proliferating cells |
| MUM1 | Plasma cells, small percentage of B and T cells, plasma cell neoplasms, lymphoplasmacytic lymphoma, some large B-cell lymphomas, other B-cell lymphomas, Hodgkin lymphoma |
| Myeloperoxidase | Myeloid cells, myeloid leukemias |
| Oct-2 | B cells including plasma cells, B-cell neoplasms including plasma cell neoplasms |
| Pax5 | B cells except plasma cells, B-cell lymphomas including B-lymphoblastic leukemia/lymphoma, Hodgkin lymphoma, small cell carcinomas and Merkel cell carcinoma |
| TDT (terminal deoxyribonucleotidyl transferase) | Precursor B, T, or NK cells, B- and T-lymphoblastic leukemia/lymphoma, blastic plasmacytoid dendritic cell neoplasm |
CUTANEOUS T-CELL AND NK-CELL LYMPHOMAS
Cutaneous T-cell lymphomas represent a heterogeneous group of neoplasms which show considerable variation in clinical presentation, histopathology, and prognosis.41. and 42.
The development of techniques to establish T-cell clonality in infiltrates in the skin has greatly contributed to the diagnosis of these disorders; they must still be interpreted in conjunction with conventional histology and immunohistochemistry.43. and 44. Rearrangement of T-cell receptor genes (TCR) α,β,γ,δ results in diversity which can be exploited by PCR and Southern blot technology to establish clonality.21.45. and 46. The majority of cutaneous T-cell lymphomas express the αβ TCR but all αβ TCR-positive cells express at least one rearrangement of the γ-chain allele and the TCR-γ gene is frequently assayed to establish clonality using a limited number of primers.43. and 44. However, it has been demonstrated that the use of a comprehensive set of primers is required to give an optimal detection rate of TCR-γ gene rearrangements. 45
It has been suggested that the detection of a clonal process in a polyclonal background is more readily detected with assays directed at the TCR-β locus. 47 Multiplex PCR assays detect virtually all clonal T-cell populations and have the added advantage of detecting γδ proliferations. 48
There are reports of complete regression of cutaneous T-cell lymphomas after biopsy. 49
KIT (CD117) expression is very rare in all types of primary cutaneous T-cell lymphoma. 50
Primary cutaneous T-cell lymphomas occurring after organ transplantation are rare and do not have the usual prognosis associated with particular subsets. 51
There are reports of cutaneous T-cell lymphoma evolving from Ofuji papuloerythroderma. 52 This entity is further discussed on page 508.
MYCOSIS FUNGOIDES AND SUBTYPES
Mycosis fungoides (MF) is a clinically and pathologically distinct form of cutaneous lymphoma characterized by an epidermotropic infiltrate of small to medium-sized T lymphocytes. In the WHO/EORTC classification, the term is reserved for those cases having classical features in which there is progression from patches to plaques to tumors (the Alibert–Bazin type) or variants which have a similar course.15. and 53.
Although it represents almost 50% of all primary cutaneous lymphomas,15. and 54. it is still uncommon. True incidence figures of MF are difficult to collate as clinical subtypes of cutaneous T-cell lymphomas (CTCL) are not clearly distinguished in many registries.54.55. and 56. There appears to be a rising incidence of CTCL in the United States. 57 There are differences in incidence among various racial groups.57.58. and 59. It usually arises in late adulthood.60.61.62.63. and 64. There is a definite male predominance. 64 It has been reported in identical twins, 65 and very rarely in families66. and 67. or spouses. 68
Lesions tend to develop on the lower part of the trunk and thighs, and on the breasts in females. 69 In advanced stages the entire body may be affected including the face and scalp. The palms and soles have been involved in some cases. MF typically has an indolent course with slow progression over years or decades. 3 In one study of progression of MF from patch stage to death from systemic spread, the overall average disease duration was 12.4 years. 70
MF is rare in children and young adults.64.71.72. and 73. Lesions in children are frequently hypopigmented, particularly in dark-skinned individuals.74. and 75. MF arising in children or young adults is not more aggressive than that appearing in adult life.72. and 74. Young patients with limited skin disease may have a slightly better disease-specific survival than older patients. 71
The etiopathogenesis of MF is unknown. There appears to be an association with particular HLA class II subtypes but not HLA class I subtypes. 76 There is no good evidence that it is linked to viruses such as HTLV-1, HHV-8, or HTLV-5.77.78.79.80.81.82.83.84.85.86.87.88. and 89. Most cases of cutaneous T-cell lymphoma in HIV/AIDS patients are erythrodermic, have a CD8+ phenotype, and are rapidly progressive. It has been suggested that MF arises in a background of chronic inflammation as a response to chronic antigen stimulation. 90 There is controversy as to the existence of a preceding stage (premycotic eruption), and its nature.91. and 92. Rare cases have been reported after solid organ transplantation. 93
Although the characteristic evolution of MF is from patch through plaque to tumor, 94 this is not invariably the case.71. and 95.
Intractable pruritus is sometimes present, occasionally preceding the appearance of skin lesions by years. 96 Histological evidence of MF has been identified in the skin of some of these cases despite the apparent absence of lesions.97. and 98.
Almost all cases of MF are a monoclonal proliferation of CD4+ cells. This has been established by PCR for TCR gene rearrangement in from 52% to 90% of cases. 34 Not all the cells in lesions are neoplastic: many are reactive. 99 Micromanipulation and laser-beam microdissection have demonstrated a monoclonal population of T cells in the epidermis in early (patch stage) lesions of MF.39. and 99. Polyclonal (reactive) T lymphocytes are more common in the dermis than are clonal T lymphocytes in these lesions. 99
Although it is classified as a primary cutaneous lymphoma, 15 circulating clonal T cells can be demonstrated in the peripheral blood even in early stages of MF by sensitive PCR techniques. 100 Clones are identical to those in the skin. 101
An MF-like picture has been associated with therapeutic ingestion of carbamazepine, 102 captopril, 103 quinine, 104 fluoxetine, 105 and phenytoin;106. and 107. the eruption cleared in each instance following cessation of the drug. 107
The patch stage consists of ill-defined patches of varying hue, often with a fine scale. They are irregular in size and shape and have a random distribution, usually on the trunk. This stage may persist for many years before progression occurs.70. and 108.
It seems appropriate to regard large plaque parapsoriasis (LPP) as early MF as 10–30% of cases progress to overt MF.91.109.110. and 111. Clonal TCR gene rearrangements have also been identified in a proportion of cases. 112 Poikiloderma atrophicans vasculare is the atrophic form of the patch stage of MF. 113 On the other hand there is less acceptance that small plaque parapsoriasis (digitate dermatosis, chronic superficial scaly dermatitis) is early MF despite views to the contrary (see p. 119).96.111.114.115. and 116. If progression does occur it is a rare event. In a recent study of 27 cases of small plaque parapsoriasis, followed for a mean of 10 years, there was apparent progression to plaque stage MF in only one case. 117
The plaque stage is characterized by well-demarcated lesions which are often annular or arciform in arrangement. They are red to violaceous in appearance and occasionally scaly. 113 The plaques may develop de novo or from patches. In the early stages, lesions are often limited to less than 10% of the skin surface, but they may be more widespread, particularly in the late plaque stage. 113
Large plaque parapsoriasis is characterized by irregular erythematous patches with minimal scale rather than plaques as the name implies. 111 Lesions occur on the trunk and major flexures and are usually larger than 6 cm in diameter.109. and 111. With time, atrophic (poikilodermatous) change may supervene in a proportion of cases. 111
Tumors usually develop in pre-existing lesions. 94 The tumors are violaceous to deep red in color, with a tense shiny surface. Ulceration may occur. The lesions usually measure 1 cm in diameter or more.
In one series which examined the progression of the disease through various stages, the average duration of the patch stage was 7.2 years, the plaque stage 2.3 years, and the tumor stage 1.8 years. 70
The term ‘d’emblee form’, used in the past to refer to cases presenting with tumors that were not preceded by patches or plaques, should no longer be used as most represent other T- and B-cell lymphomas which present with tumors.42. and 118.
There is a long list of non-classic presentations of MF64.119. and 120. which although not the typical Alibert–Bazin type have a behavior similar to classic MF. These include hypopigmented lesions,121.122.123.124.125.126.127.128. and 129. hyperpigmented lesions,87. and 130. leukoderma, 131 bullae,132.133.134.135.136.137. and 138. dyshidrotic lesions, 139 perioral dermatitis-like lesions, 140 palmar-plantar lesions,141.142. and 143. papules,144.145. and 146. pustules, acneiform, hyperkeratotic, verrucous,147.148. and 149. poikilodermatous, 150 anetodermic, 151 annular erythema, 152 granuloma annulare-like, 153 pyoderma gangrenosum-like, 154 and plaques resembling acanthosis nigricans,155. and 156. or keratosis lichenoides chronica. 157
At present, so-called ‘syringotropic MF’ is not regarded as a separate entity, unlike folliculotropic MF.64.158. and 159. By itself, it may present with punctate erythema. 160 Syringotropism can be seen in otherwise classic MF and in folliculotropic MF.160.161.162. and 163. In many cases syringolymphoid hyperplasia, characterized clinically by scaly plaques associated with hair loss and anhidrosis, represents a T-cell lymphoma.164. and 165. Rarely there may be erythroderma. 166
Acquired epidermal cysts, 167 nail dystrophy,167. and 168. neutrophilic dermatoses, 169 and acquired ichthyosis170.171. and 172. have been reported in association with MF. Second neoplasms including skin cancers, other lymphomas, and internal malignancies have also been reported, some apparently related to therapy.173.174.175. and 176. Oral lesions occur rarely.177. and 178.
MF-associated mucinosis is now classified separately as a variant of MF with folliculotropic MF and is discussed separately.
MF may also present as purpuric lesions (see p. 232) which resemble or are indistinguishable from the pigmented purpuric dermatoses (PPD).179.180.181. and 182. Clinical suspicion should be aroused when the distribution of purpuric lesions is unusual for one of the forms of PPD, or lesions are extensive, long-standing and have a reticular pattern. 179 The separation of the two conditions is not always possible by routine histology or even with TCR gene rearrangement studies. 180
Solitary lesions with the histopathology of early MF, and distinct from pagetoid reticulosis, have been reported,183.184.185.186. and 187. sometimes in unusual sites (penis). 188 In one small series of such cases, clonal TCR gene rearrangement was found in 50% of cases. No evidence of progression to classic MF was observed in any of the cases. 184 It is not clear if these lesions represent true lesions of MF or are simulants. 189
Erythroderma can develop at any stage in the evolution of MF. 94 The distinction between Sézary syndrome (SS) and erythrodermic MF is based on the absence of significant numbers of circulating ‘Sézary cells’ in the peripheral blood as defined below. When hematologic findings fulfill the criteria for Sézary syndrome the condition is classified as ‘secondary SS’ or ‘SS preceded by MF’. 190
Circulating T lymphocytes exhibiting the same clonal TCR gene rearrangements as those in skin lesions can be found even in early stages of MF. 101
Involvement of lymph nodes and other organs may occur in late stages of the disease. 191 Lymphadenopathy in the early stages is due in most cases to dermatopathic lymphadenopathy, a reactive condition that may be seen in lymph nodes draining skin affected by a variety of inflammatory dermatoses as well as MF. 192 However, sensitive PCR techniques have shown involvement of lymph nodes by clonal T lymphocytes even at early stages of MF.193.194. and 195. Molecular evaluation of lymph nodes for staging purposes has been investigated,196. and 197. but detection of a monoclonal population of T cells by PCR of lymph nodes does not appear to be superior to clinical and histological examination of lymph nodes in predicting clinical outcomes. 198 Grading of more than one lymph node for staging purposes is reported to be more accurate than for a single node. 199 The detection of clonality by Southern blot techniques may be more useful in predicting outcome than using more sensitive PCR techniques. 200 One study concluded that detection of a monoclonal population of T cells in lymph nodes by Southern blot analysis194 or by more sensitive PCR techniques201 was predictive of a poor clinical outcome and reduced probability of survival. Detection of a monoclonal population in peripheral blood is also an adverse feature. 202 Visceral involvement is frequently found at autopsy. The lung, spleen, liver, and kidney are most frequently involved but every organ can be infiltrated by tumor cells.203.204.205.206. and 207.
There have been numerous studies on the prognosis and survival in patients with MF.208.209.210.211.212.213.214. and 215. Different and developing treatment modalities would be expected to alter the prognosis of MF in the future.
The clinical course of MF is quite variable; in the majority of cases it is indolent. Spontaneous resolution of individual lesions may occur at any time during the disease. The estimated 5-year survival of patients in the EORTC series was 87%. 3 Patients are staged T1 to T4 (T1 = limited patch/plaque, T2 = generalized patch/plaques, T3 = tumor, T4 = erythroderma). Patients with stage T1 disease have a very favorable outcome; their life expectancy is not altered by the disease. 216 In a recent study of long-term outcome for patients with MF and Sézary syndrome, age, stage (T classification), and the presence of extracutaneous disease were important predictors of survival. 214 Development of extracutaneous lymphoma has been reported in cases of late-stage MF treated with bexarotene. 217 Transformation to a large cell lymphoma218 is a bad prognostic feature.219.220.221. and 222. In one series, those cases with a CD30+ large cell transformation had a better prognosis that those with a CD30− phenotype. 221 CD30+ large cell transformation has been reported after PUVA, and alefacept therapy for MF. 223 Second B-cell lymphomas have been reported in patients with pre-existing MF. 224
Serum thymus and activation-regulated chemokine (TARC/CCL17) levels are useful for assessing the disease activity in patients with MF. 225
The outcome of patients with juvenile-onset MF is similar to that of adult-onset disease. 73 Pregnancy appears to have no impact on the course of early stage MF. 226 PCR for clonal T-cell rearrangements has identified residual disease in a third of patients with complete clinical remission. 227
Histopathology
There is considerable literature on the histological diagnosis of mycosis fungoides, particularly in the early stages.228. and 229. A combination of routine histopathology, immunohistochemistry, and gene rearrangement studies is now possible. All have their limits. Attempts have been made to ‘weigh’ the importance of various histological parameters in arriving at a diagnosis.230. and 231. Multiple biopsies over a period of time are often needed for a diagnosis. Biopsy appearances can be altered by therapies such as corticosteroids and UVA therapy. Many studies have shown that interobserver agreement on the histological diagnosis of MF is not high. 232 Despite the use of other techniques, some authors maintain that the diagnosis of early MF still depends on clinical features and conventional histology and recommend further biopsies where there are no clear-cut diagnostic features in a biopsy. 233
Patch stage
In the patch stage of MF a combination of architectural and cytological features is used to make the diagnosis. This includes changes in the epidermis and dermis. In the earliest stage there is a relatively sparse infiltrate of lymphocytes spread along the slightly expanded papillary dermis with little tendency to aggregate around vessels of the superficial plexus. Within the epidermis, lymphocytes are typically confined to the basal layers of the epidermis, either as single cells in a ‘string of beads’ arrangement or as small groups of cells (Fig. 41.1). These cells are often surrounded by a clear ‘halo’, a ‘meaningful artifact’ not caused by mucin accumulation. 234 There is usually little or no spongiosis.235. and 236. The Pautrier microabscess (sharply marginated discrete clusters of lymphocytes in close apposition with one another, within the epidermis) is, when strictly defined, highly characteristic of MF (Fig. 41.2). They are uncommon in the patch stage, however, and if this feature is given undue importance many cases of MF will be missed.237. and 238. Occasionally the histological appearance mimics a melanocytic lesion. Atypia of cells may be minimal in earlier stages of MF but in some cases, the epidermal lymphocytes are larger that those in the dermis. 92 Cytological abnormalities may be seen in thin plastic sections that are not apparent in conventional sections. 103 There is frequently, but not invariably fibrosis in the papillary dermis in the form of haphazardly arranged wiry collagen bundles.92. and 103. The epidermis may show mild acanthosis; in poikilodermatous and atrophic lesions the epidermis is thin. 235 Necrosis of the epidermis is occasionally present. 228 Basal vacuolar change and pigment incontinence are present in these lesions. 239 In some cases there is a marked lichenoid reaction with histology resembling lichen planus. Unlike lichen planus there may be plasma cells and eosinophils in the dermal infiltrate as well as atypical lymphocytes. Infiltration of eccrine sweat duct structures may be seen in patch and other stages of MF and may remain after therapy.240. and 241.
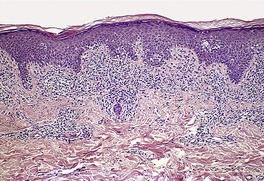 |
| Fig. 41.1 Mycosis fungoides. There is a band-like dermal infiltrate with atypical lymphocytes in the basal epidermis. (H & E) |
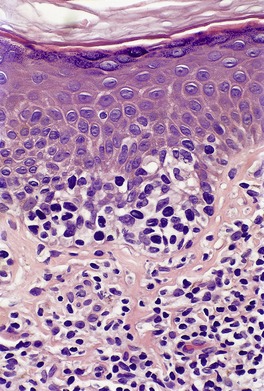 |
| Fig. 41.2 Mycosis fungoides. A collection of epidermal atypical lymphocytes forming a Pautrier microabscess. (H & E) |
The diagnostic changes in large plaque parapsoriasis may be subtle with epidermal changes of mild psoriasiform hyperplasia, overlying mild orthokeratosis and spotty parakeratosis, and a sparse dermal cellular infiltrate. The dermal infiltrate may extend upward into and fill the dermal papillae. Pautrier microabscesses are not seen and cellular atypia is often minimal. 242 The atrophic lesions have identical changes to other atrophic lesions of MF. 243
The pigmented purpuric dermatoses (PPD) and pigmented purpuric dermatitis-like lesions of MF may have similar histological appearances. In PPD-like MF there may be features typical of MF, but some cases have similar features to PPD. Both PPD and PPD-like MF may have lymphocytes aligned in the basal layer of the epidermis and occasional Civatte bodies. Dermal edema is more common in PPD. Atypia of lymphocytes is seen in MF-like PPD and lymphocytes may be seen in upper levels of the epidermis. 180 PCR studies may show clonal rearrangements of TCR genes in lichenoid forms of PPD, making distinction of some cases from MF difficult. 180
Plaque stage
Patch and plaque stages are part of a progression. In plaques of MF, the infiltrate is more dense and atypical lymphocytes are more common. The lymphocytes measure 10–30 µm in diameter and their nuclei are often obviously indented, ‘prune-like’ or cerebriform. Prominent convolutions are best appreciated in thin sections. Nuclear morphometry of CD3+ T cells has been used as a diagnostic technique to distinguish neoplastic T lymphocytes from reactive ones. 244 It has been suggested that it is not possible to distinguish MF from spongiotic dermatitis based on identifying lymphocyte atypia alone in routine sections. 245
Small collections of cells may aggregate around vessels of the superficial plexus and less often the deep plexus. They also extend round adnexae, particularly pilosebaceous follicles. 246
In addition to lymphocytes, the infiltrate usually contains a small number of eosinophils and sometimes plasma cells. 237 Epidermotropism is still a conspicuous feature. Pautrier microabscesses are seen in more than 50% of biopsies; this proportion increases if step sections are examined. Epidermal changes include parakeratosis, mild psoriasiform hyperplasia, and epidermal mucinosis.237. and 247. Mild spongiosis does not exclude the diagnosis of MF as sometimes claimed, 235 but spongiotic microvesiculation is rare. 248 Spongiotic foci resembling Pautrier microabscesses (pseudo-Pautrier microabscesses) are sometimes seen in spongiotic processes such as allergic contact dermatitis and pityriasis rosea. 249 These foci contain monocyte-like cells, Langerhans cells, and rare lymphocytes. Cell nuclei are pale and less complex than in the component cells of true Pautrier microabscesses. These structures are often vase-shaped and appear to open onto the epidermal surface. They may also be seen in MF.250. and 251. Biopsies of apparently normal skin in patients with plaque stage MF sometimes show a mild superficial perivascular infiltrate of lymphocytes, often with epidermotropism. 252
Tumor stage
In the tumor stage, the infiltrate has a more monomorphic appearance and is dominated by atypical cells. 253 The proportion of tumor cells relative to reactive cells increases. Mitotic figures are easily seen. The entire dermis is often involved and extension into subcutis may occur. Deep dermal and subcutaneous nodules are particularly likely to occur if electron beam therapy has been given to a pre-existing lesion in the same region. 254 Epidermotropism and Pautrier microabscesses are uncommon in the tumor stage. Transformation to a diffuse large cell lymphoma may occur. This was defined in one series by the presence of lymphocytes >4 times the size of small lymphocytes in more than 25% of the infiltrate or microscopic nodules of the same. 222 The cells can have variable features and resemble the cells seen in anaplastic large cell lymphoma, or resemble immunoblasts or large pleomorphic cells.
Syringotropism may be the predominant pattern of infiltration with invasion of components of the eccrine coil and duct sometimes associated with proliferation of the epithelial structures. 160
The separation of MF from certain inflammatory dermatoses is not as clear cut as has been suggested. 92 There is often disagreement between pathologists in individual cases. Various attempts have been made to provide diagnostic principles, to weigh the importance of particular features and to standardize reporting of cases.255.256.257.258. and 259. The important histological features in most studies appear to be:
Histology is not useful in predicting disease course. 260
Granulomas are a rare finding in mycosis fungoides (granulomatous MF).248.261.262.263.264.265.266.267.268.269.270. and 271. They are usually small and tuberculoid in type but they may be palisaded and mimic granuloma annulare. 272 At other times the granulomas are poorly formed and consist of small collections of multinucleate histiocytes. The granulomas are more localized than in granulomatous slack skin. Granulomas in MF should be distinguished from small collections of lipidized macrophages (dystrophic xanthomatosis) which are rare findings in the dermis in MF.262.273. and 274. A variant with interstitial lymphoid infiltrates superficially resembling granuloma annulare or inflammatory morphea has been reported,275. and 276. as has generalized granuloma annulare associated with granulomatous MF. 277 Rarely there is extensive fibrosis and mucin in the dermis (fibromucinous variant).248. and 278.
Other rare histological changes include the presence of vasculitis, 279 and the formation of bullae. In the bullous lesions the split may occur at any level. The majority of cases have been subepidermal in location with negative immunofluorescence. 132 Signet-ring cells constituted the infiltrate in one case. 280 In hypopigmented lesions of MF, there is a reduction in melanin in the basal cell layer and, sometimes, melanin incontinence. 124
Epidermal changes such as mild epidermal hyperplasia, dyskeratotic cells, and atypical keratinocytes with large nuclei may be seen following topical treatment with nitrogen mustard. 281
Histological mimics of MF have been reviewed. 282
Immunohistochemistry and cytogenetics
The tumor cells are typically CD3+, CD4+, CD45R0+, and usually CD8−, CD30−.283.284.285. and 286. The cells have the characteristics of mature memory cells of the Th2 subset. 287 There is variable expression of the T-cell markers CD2, CD5, and CD7.
Tumor cells frequently lack expression of CD7.288. and 289. Although this aberrant feature was thought to be specific for a lymphomatous T-cell process and a useful feature in the differential diagnosis of reactive from neoplastic processes, it has been reported in inflammatory conditions.290. and 291. Loss of several T-cell markers is unusual in reactive processes and would favor lymphoma.292. and 293. Loss of the T-cell marker CD62L has also been used as evidence for a neoplastic T-cell proliferation, 294 as has a T-cell proliferation which is CD45RB+, CD45RO−. 295 In the tumor stage there is commonly an aberrant phenotype with loss of T-cell antigens.296.297.298. and 299. Rare cases with similar behavior to classic MF have been reported with a CD4−, CD8+ phenotype or CD4−, CD8–.300.301.302. and 303. Phenotypic shift from CD4+ to CD8+ has been reported. Cases of hypopigmented MF are particularly likely to have a CD8+ phenotype.128. and 129.
Rare CD4−, CD8+ cases are CD56+. 304 Reactive CD8+ cells can also be found in the lesions of MF. 305 Cytotoxic proteins (TIA-1, granzyme B) are sometimes expressed by the CD4+ cells, particularly in late stages of the disease. 292 With progression of the disease some cases may transform to a large cell lymphoma which may be CD30+ (secondary large cell anaplastic lymphoma) or CD30–.219.222.306. and 307. Such CD30+ tumors are not associated with the t(2;5) translocation. 308 Rarely CD30+ cells are found in appreciable numbers in the patch stage of MF. 309 MUM1, a marker for plasma cells, late B cells, and activated T cells, is expressed in transformed CD30+ cells. 310 Expression of certain CD44 splice variants may be linked to systemic spread.311. and 312. In addition to lymphocytes, the dermal infiltrate in MF includes numerous dendritic cells that express CD1a or factor XIIIa.313.314. and 315. The T cells within the epidermis usually express proliferating cell nuclear antigen (PCNA). Small B lymphocytes are sometimes present in the dermis in small numbers. Rarely neoplastic T cells co-express CD20. 316 Malignant T cells express CCR4 (CC chemokine receptor 4).225. and 271.
In most cases, neoplastic lymphocytes of MF express the αβ TCR but rare cases have been reported in which the γδ receptors are expressed.317. and 318. There are many studies in which the detection of clonal TCR gene rearrangements has been used in the diagnosis of MF, particularly in the early, histologically equivocal lesions of MF.34.288.319.320.321.322.323. and 324. The significance of demonstrable clonal TCR gene rearrangements in suspect lesions remains controversial to a certain extent but most authors agree that such a finding warrants clinical monitoring. Many such cases have progressed to frank MF. 34 TCR analysis from two different sites has high specificity in distinguishing MF from inflammatory dermatoses. 325 Unfortunately, early MF is not uncommonly negative for clonal TCR rearrangements. 230
Expression of the Fli-1 transcription factor is associated with progression to tumor stage MF. 326
Electron microscopy
There is a great diversity in the morphology of the atypical lymphocytes in MF. The characteristic cell has a highly convoluted (cerebriform) nucleus with heterochromatin located predominantly beneath the nuclear membrane (Fig. 41.3). The cytoplasm contains multivesicular bodies and mitochondria, which are sometimes clumped. Some authors use the term ‘Sézary cell’ for a small to medium-sized lymphocyte with a highly convoluted nucleus and the term ‘mycosis cell’ for a cell which is slightly larger with fewer nuclear indentations.333. and 334. Most authors, however, use the terms, ‘Sézary cell’, ‘mycosis cell’, and ‘Lutzner cell’ interchangeably for the various atypical cells, recognizing that intermediate forms also exist.108. and 335.
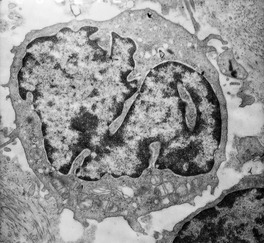 |
| Fig. 41.3 Mycosis fungoides. A mycosis cell with its characteristic indented nucleus. (×12 500) |
A much larger cell, sometimes called the ‘pleomorphic cell’, is seen in the tumor stage. It has a vesicular nucleus and a conspicuous nucleolus. 334 The nucleus of the pleomorphic cell may be variably convoluted.
Folliculotropic mycosis fungoides
Folliculotropic MF has been classified as a separate entity to classic MF in the WHO/EORTC classification because it has distinctive clinical and histological features, is more resistant to standard therapies, and has a worse prognosis.15.337. and 338.
The term incorporates pilotropic MF, follicular MF, and MF-associated mucinosis. 15 It has also been reported as follicular cutaneous T-cell lymphoma. 339 In this condition, the neoplastic T-cell infiltrate is predominantly folliculocentric and epidermal involvement is absent or minimal.
This form occurs predominantly in adults but there is a broad age range including children. 337 Males are affected more commonly than females. It has been reported in association with lithium therapy. 340
The head and neck are the sites most commonly involved. 338 Lesions range from grouped folliculocentric papules, to plaques and tumors. There is sometimes associated alopecia, cysts, and comedones. It may be associated with nail involvement and ungual mucinosis. 341 Rarely, there is erythroderma or Sézary syndrome.342. and 343. Pruritus is common and may be severe. 337 As a result of follicular mucinosis, there is mucinorrhea in some cases.
The prognosis of this group is generally worse than classic MF. There may be transformation to a CD30+ large cell lymphoma.344. and 345. Spread to extracutaneous sites has also been reported.337. and 346. Sometimes the progress of the lymphoma is rapid. 347 In one large series, the overall survival at 5 years and 10 years was 64% and 14%, respectively. 337 In a recent series, the 10-year survival was 82% and the 15-year survival 41%. 338
Histopathology337
There is a follicular and perifollicular infiltrate of small to medium-sized lymphocytes with or without follicular mucin (Fig. 41.4).348. and 349. The nuclei are often conspicuously cerebriform. The infiltrate may also be present around vessels and the eccrine apparatus, sometimes extending into eccrine epithelium in a similar manner to that in the follicle. Mucin may be minimal or form small pools in the follicular epithelium. This can be highlighted by Alcian blue stains. Pautrier microabscesses are occasionally present. 350 Involvement of the epidermis is not present or is minimal.
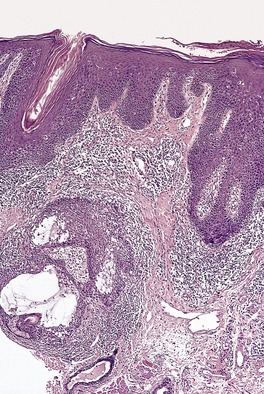 |
| Fig. 41.4 Mycosis fungoides-associated follicular mucinosis. (H & E) |
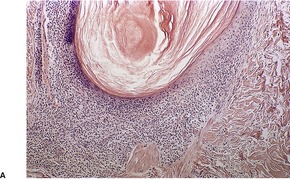
Follicular dilatation and follicular cyst formation may be evident (Fig. 41.5). A granulomatous reaction to ruptured follicular epithelium is seen occasionally.351.352. and 353.
 |
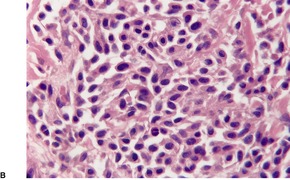 |
| Fig. 41.5 Pilotropic (follicular) mycosis fungoides. (A) Dilated follicle with atypical lymphoid infiltrate. (B) There is a follicular infiltrate of cerebriform T cells. (H & E) |
Sometimes there is marked basaloid follicular epithelial hyperplasia, ‘basaloid folliculolymphoid hyperplasia’, 354 or trichilemmal follicular hyperplasia. 355 A similar proliferation of eccrine epithelium is sometimes seen and there is overlap with so-called ‘syringolymphoid hyperplasia’, many cases of which represent a T-cell lymphoma. Inflammatory cells including eosinophils and plasma cells are commonly seen in the infiltrates. 356 Small numbers of B cells are also present; rarely they may be in such number as to mimic a B-cell neoplasm. 357 Sometimes there is an interstitial dermatitis-like pattern. 353
In more advanced cases there may be more confluent infiltration of the dermis and larger atypical cells are more common.
Immunohistochemistry and cytogenetics
The atypical lymphocytes have CD3+, CD4+, CD8− phenotype. Scattered large atypical CD30+ or CD30− cells are commonly seen and they may become more confluent in large cell transformation. It has been suggested that the involvement of hair follicles is related to the overexpression of intercellular adhesion molecule-1(ICAM-1) by keratinocytes in the hair follicle. 358
Pagetoid reticulosis
Pagetoid reticulosis (Woringer–Kolopp disease)359. and 360. is a rare distinct variant of MF which presents clinically as large, usually solitary, slow growing, erythematous, scaly or verrucous patches or plaques.361.362. and 363. Lesions are typically found on the distal part of the limbs. It has been described on the penis. 364 Adult males are predominantly affected. Cases in childhood are extremely rare. 365
A prolonged disease-free survival after simple excision or local irradiation is usual. Extracutaneous dissemination has not been reported. 3
Ketron-Goodman disease, which has widely disseminated lesions and a different clinical course, is now regarded as a separate disorder, either a variant of MF or primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma (see p. 987).15.366. and 367.
Histopathology
The epidermis is characteristically infiltrated by large atypical mononuclear cells with pale eosinophilic cytoplasm, a large nucleus and prominent nucleolus (Fig. 41.6).368. and 369. Cells are arranged singly or in nests or clusters. This produces a pattern resembling Paget’s disease or melanoma. Atypical cells are present at all levels of the epidermis but are most prominent in the lower third. 361 Cells in the upper layers of the epidermis may show subtle degenerative changes. 368 There are scattered mitotic figures. The epidermis is markedly acanthotic with overlying hyperkeratosis and patchy parakeratosis.
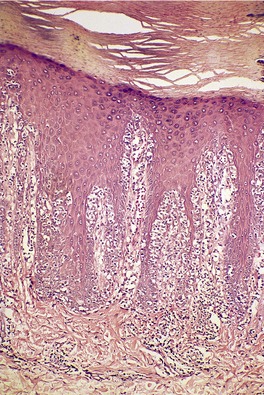 |
| Fig. 41.6 Pagetoid reticulosis. There is marked epidermotropism of atypical lymphocytes. (H & E) |
Unlike MF, there are no atypical cells in the dermis, which contains a dense, banal infiltrate of small lymphocytes, histiocytes, and some plasma cells. 368 There are usually no eosinophils.
Immunohistochemistry and cytogenetics
Tumor lymphocytes have a CD3+, CD4+, CD8− or CD3+, CD4−, CD8+ immunophenotype. CD30 is sometimes expressed.362.370.371. and 372. Clonal rearrangements of TCR genes have been demonstrated. This is usually an αβ TCR gene rearrangement but rarely may be an γδ rearrangement.371.373. and 374. Neoplastic cells have been reported to express cutaneous lymphocytic antigen (HECA 452), a skin homing receptor, which might explain the exquisite epidermotropism of this lesion. 371
Granulomatous slack skin
Granulomatous slack skin is a very rare cutaneous lymphoma in which pendulous folds of skin develop in large pre-existing erythematous plaques.377.378.379. and 380. There is predilection for flexural areas, particularly the axilla and groin. 381 There is a clonal infiltrate of CD4+ T cells in the dermis associated with granulomatous inflammation. This condition occurs predominantly in males. 3 Most patients have an indolent course although in a third of patients there is an association with Hodgkin lymphoma.262.377.382. and 383. It may also be associated with classical MF. 380 Another complication reported in 2007, large cell transformation, resulted in the death of the affected child. 384 Recurrence of the pendulous skin folds after surgical excision has been reported. 377 Rarely there is evidence of extracutaneous lymph node involvement. 385
Histopathology377
Early lesions exhibit a superficial, or superficial and deep perivascular lymphocytoid infiltrate, psoriasiform epidermal hyperplasia, slight spongiosis, parakeratosis, and occasional lymphocytes in the lower half of the epidermis. Occasional multinucleate histiocytes are seen in the dermal infiltrate. In established lesions, there is permeation of the entire dermis and the subcutis by a dense infiltrate of small lymphocytes with convoluted nuclei. Characteristically, there are many multinucleate giant cells scattered uniformly throughout the lymphocytic infiltrate. These giant cells contain large numbers of nuclei. Their cytoplasm may contain lymphocytes and elastic fibers. Cellular infiltrates in the upper dermis may be band-like. The dermis between the cellular aggregates is markedly edematous or fibrotic.
Stains for elastic tissue show a complete absence of elastic fibers from the dermis. Occasionally, calcified elastic fibers are seen.378. and 386. The epidermis is infiltrated by lymphocytes, either singly or in small clusters in a pattern similar to the patch or plaque stage of MF.
Immunohistochemistry and cytogenetics
The small lymphoid cells are CD3+, CD4+ T cells. The multinucleate cells express histiocyte markers such as CD68.387. and 388. Rarely, large CD30+ cells are part of the infiltrate. 389 The neoplastic cells are generally αβ T lymphocytes and clonal TCR rearrangements of the β-chain have been demonstrated.379.386. and 390. Occasional cases demonstrate a clonal γ-chain rearrangement, 385 or do not demonstrate a clonal rearrangement. 391 A translocation t(3;9)(q12;24) has been demonstrated in one case. 392
SÉZARY SYNDROME
Sézary syndrome (SS) is an uncommon form of cutaneous T-cell lymphoma. In the United States, the incidence is in the vicinity of 0.3 per 106 persons. 57 In the current WHO/EORTC classification it is designated as an entity distinct from mycosis fungoides, although some would regard it as a manifestation of mycosis fungoides, 393 or a leukemic stage of mycosis fungoides. 394
The International Society for Cutaneous Lymphomas (ISCL) recognizes several variants of erythrodermic cutaneous T-cell lymphoma, erythroderma being defined as diffuse erythema involving 80% of the skin surface. Within this spectrum is SS which may arise de novo or following mycosis fungoides, erythrodermic mycosis fungoides which lacks the hematologic findings of SS, and erythrodermic cutaneous T-cell lymphoma, not otherwise specified. 190
Sézary syndrome was defined historically by the triad of erythroderma, lymphadenopathy, and circulating atypical mononuclear cells in the peripheral blood. 395 Lymphadenopathy, although common in SS, is no longer considered requisite for the diagnosis.
The circulating atypical T cells with hyperconvoluted ‘cerebriform’ nuclei (a nuclear contour index of 6.5 or more) have been called ‘Sézary’ or ‘Lutzner’ cells: there are small and large variants. 393 They can be recognized in blood films stained by the Giemsa or Wright methods but cell counts using smears may underestimate numbers. 396 These cells are not specific for SS and may be seen in small numbers in other cutaneous inflammatory conditions and in normal blood. 190
The ISCL defines SS as generalized erythroderma with hematologic criteria which include: an absolute Sézary cell count of at least 1000 cells/mm3; a CD4/CD8 ratio of 10 or more; aberrant expression of pan T-cell markers CD2, CD3, CD4, CD5; increased lymphocyte counts with evidence of T-cell clonality in the blood and a chromosomally abnormal T-cell clone. 190
Russell-Jones has developed an algorithm for the diagnostic separation of erythrodermic T-cell lymphoma and non-neoplastic causes of erythroderma. 397
Less constant clinical features include cutaneous edema, alopecia, nail dystrophy, and palmar and plantar keratoderma. 398 Rarely bullous lesions, 399 plane xanthomas, 400 vitiligo, 401 leukoderma, 402 or a monoclonal gammopathy403 may develop. Peripheral blood eosinophilia is present in many cases.55. and 404. Increased blood immunoglobulin levels are also common. 405
Prognostic parameters are still being investigated, but visceral involvement, advanced age, long interval before diagnosis, previous history of mycosis fungoides, and circulating Sézary cell counts of more than 5% of the total lymphocyte count, and the presence of the Epstein–Barr virus (EBV) genome in keratinocytes are unfavorable prognostic features.394.406. and 407. In one series the 5-year survival was 33.5%. 406 Death is frequently due to systemic opportunistic infections as a result of immunosuppression. 408
Sézary syndrome usually develops in late adult life. Cases in childhood are extremely rare. 409 It may be preceded by another chronic inflammatory dermatosis such as atopic dermatitis. 410 One recent study found no causal relationship between atopy and SS or MF, however. 411 It has been reported in association with infliximab therapy, 412 and following exposure to ionizing radiation. 413 Development of typical lesions of mycosis fungoides has been reported after remission of primary SS, 414 and secondary SS may follow folliculotropic mycosis fungoides. 415 Large cell transformation in skin, lymph nodes, and peripheral blood has also been reported.416. and 417. There is an increased risk of squamous cell carcinoma of the skin, Hodgkin lymphoma, and internal malignancies in patients with SS.175.418. and 419.
Some cases of erythroderma have demonstrable T-cell clones in blood without fulfilling the criteria for SS or erythrodermic MF. 420
Histopathology400.421.422. and 423.
There is great variability in the histological findings. In many cases they are similar to those in MF.3. and 424. The same problems of specificity apply as with the early stages of mycosis fungoides discussed above. In up to 40% of cases, however, the biopsy appearances are non-diagnostic.425. and 426. Multiple biopsies may be more successful in obtaining a histological diagnosis of SS. Topical and other therapy often mask or obliterate diagnostic changes. Non-specific histological findings are common in SS even when a circulating T-cell clone is present in blood. 427 In one author’s experience, the diagnosis of SS could be established with greater certainty by hematologic studies rather than by skin biopsies. 426
In the most frequently observed histological patterns in SS, there is a perivascular or less often a band-like cellular infiltrate involving the papillary dermis and sometimes the upper reticular dermis as well. 422 Epidermotropism is present in some of these cases and Pautrier abscesses may be found, particularly if multiple sections are examined. 422 In one recent series epidermotropism was minimal or absent in 61% of cases. 423 The infiltrate is of varying density and is composed of small lymphocytes admixed with some larger cells with indented or cerebriform nuclei. Lymphocytic cellular atypia may be minimal or inapparent. 423 Atypical lymphocytes can be seen in spongiotic processes, 56 and some fixation protocols can produce apparent nuclear atypia. 423 The use of ultrathin sections enhances the detection of these cells. The cellular infiltrate is present in a background of variable epidermal changes, which include irregular acanthosis of the epidermis with orthokeratosis and focal parakeratosis. Spongiosis is sometimes present although usually mild. 421 The papillary dermis is fibrotic with thickened collagen bundles and scattered melanophages. Eosinophils and plasma cells may be found in small numbers. Prominent ectasia of papillary dermal vessels is present in many cases and is more pronounced than in patch/plaque lesions of MF. 428
Rarely, non-caseating granulomas may be seen in the dermal infiltrate, sometimes after therapy. 429
Immunohistochemistry and cytogenetics
The neoplastic lymphocytes in the skin and blood are CD3+, CD4+, CD45RO+, CD8−, CD30−, and frequently CD2−, CD7−. Studies employing double staining for CD4/CD8 ratios >10 in skin biopsies have been deemed unhelpful in making a diagnosis. 430 Circulating clonal CD4+ T cells have been shown to express the skin and lymph node-homing chemokine receptors CCR4, CCR10, and CCR7. Clonal TCR gene rearrangements are present in most cases and are critical if histology and immunohistochemistry are unhelpful. Analysis for TCR gene rearrangements either by PCR426 or Southern blot techniques may be difficult to perform in skin biopsies because of the small number of cells in the infiltrate and problems in extracting cells from skin. Demonstration of clonality in circulating T cells in the peripheral blood aids in the differentiation of SS from other non-neoplastic forms of erythroderma. 431 The presence of the same T-cell clone in skin and blood is strong evidence of SS in the absence of diagnostic histological changes.432. and 433. Flow cytometric counts of CD4+/CD7− cells in peripheral blood have shown a significant correlation with the number of circulating Sézary cells. 434 In some cases, however, the cells are CD4+/CD7+ which obscures the phenotypic distinction between neoplastic and normal T cells.406. and 435. Diminished expression of CD3 and lack of CD26 expression have been used to detect and enumerate atypical cells in the blood in SS.435.436. and 437.
A case of Sézary syndrome-like erythrodermic cutaneous T-cell lymphoma with a CD8+, CD56+ type phenotype has been described. 438
Electron microscopy
The Sézary cells have a convoluted nucleus with deep and narrow indentations. The cytoplasm has a number of fibrils. Ultrastructural morphometry has been used to distinguish Sézary cells in the blood from normal and reactive lymphocytes although this technique is rarely used in routine practice. 441
ADULT T-CELL LEUKEMIA/LYMPHOMA
Adult T-cell leukemia/lymphoma (ATLL) is a T-cell lymphoma resulting from infection with human T-cell lymphotrophic virus type 1 (HTLV-1), a type C retrovirus.442.443. and 444.
The virus is endemic in south-western Japan and the Caribbean as well as certain parts of South America and Africa. It has also been reported sporadically in parts of Europe and the south-eastern USA.445.446.447.448. and 449. HTLV-1 is transmitted by sexual contact, breastfeeding, infected blood products, and percutaneously.445. and 448. Familial clustering has been described. 449 The average age of onset of ATLL in a large series from Japan was 57 years (24–92 years). 450 It is very rare in children. Immigrant studies suggest there is a long latent period from viral infection to overt ATLL. 451
Several clinical forms are recognized; they have been divided into four groups by Shimoyama: 450 (1) an aggressive acute form seen in approximately 65% of patients and associated with a very high white-cell count, hepatosplenomegaly, hypercalcemia, and lytic bone lesions; (2) a chronic form associated with lower white-cell counts and no hypercalcemia. Lymphadenopathy, hepatosplenomegaly may be present as well as skin manifestations; (3) a smoldering form with normal lymphocyte counts in blood but with 1–5% ATLL cells. Skin and lung infiltrates may be present; (4) a lymphomatous form with no lymphocytosis, less than 1% circulating ATLL cells and isolated lymphadenopathy or extranodal tumors. A purely cutaneous form which resembles MF has also been described.450. and 452. The integration pattern of HTLV-1 proviral DNA may be the explanation for the heterogeneity in the behavior of ATLL.453. and 454.
Skin manifestations are often the initial manifestation of ATLL (50–70% of cases); they are found in all forms of the disease.455. and 456. Lesions are often widespread and have many forms including erythematous patches, plaques, papules, and tumors. Erythroderma and vesiculobullous and purpuric eruptions are less common cutaneous manifestations.457.458. and 459. Presentation as an isolated single skin nodule is very uncommon. 460 Non-specific skin lesions or prurigo may precede the onset of acute ATLL.461. and 462.
The prognosis depends on the clinical subtype; it is worse in the acute and lymphomatous variants with survival of 2 weeks to 1 year. 15
Atypical lymphocytes are found in the blood in 50–80% of patients at presentation; virtually all develop a leukemic phase eventually. 463
Histopathology
The cutaneous lesions of ATLL share some features with MF in having an infiltrate of atypical lymphocytes in the upper dermis with variable epidermotropism and occasional Pautrier microabscesses. Unlike MF, these microabscesses may contain prominent apoptotic fragments (Fig. 41.7). In the nodule and tumor stage the dermal infiltrate is more extensive, confluent and atypical and can extend into the subcutis. As in the tumor stage of MF, epidermotropism is less apparent. 457 In some cases the infiltrate conforms to a papular outline, an uncommon pattern in MF. 103
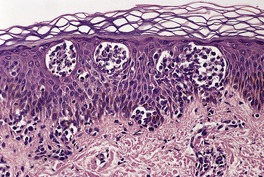 |
| Fig. 41.7 Adult T-cell leukemia/lymphoma. There is extensive epidermotropism with Pautrier microabscess formation. Characteristic apoptotic debris is just visible. (H & E) (Case kindly supplied by Dr Tetsunori Kimura, Section of Dermatology, Sapporo General Hospital, Sapporo, Japan) |
The infiltrating cells may be medium to large size, sometimes with pleomorphic or polylobated nuclei.445. and 455. Other elements, such as small lymphocytes, histiocytes, eosinophils, and plasma cells, are less common than in MF. Sometimes dermal aggregates form, resembling granulomas.464. and 465.
Atypical lymphocytes are sometimes seen in the lumina of blood vessels in the dermis. Angiocentric and angiodestructive lesions are rare manifestations of ATLL. 456
Immunohistochemistry and cytogenetics
The neoplastic cells are T lymphocytes exhibiting clonal TCR rearrangements. They are usually CD2+, CD3+, and CD4+. Rarely, the cells express a CD4+, CD8+466 or CD4−, CD8− phenotype. 459 They also express CD25, which is a distinguishing feature between ATLL and Sézary syndrome.467. and 468. HTLV-1 is clonally integrated in T cells in all cases and can be demonstrated in paraffin-embedded tissue. 469
Electron microscopy455
The tumor cells show slight to marked nuclear irregularity with a convoluted shape and a speckled chromatin pattern. The circulating lymphocytes have been termed ‘flower cells’. 449 The degree of nuclear indentation is much less than in the cells of MF and SS. The cells in ATLL have lysosomal granules and glycogen in their cytoplasm.
PRIMARY CUTANEOUS CD30+ LYMPHOPROLIFERATIVE DISORDERS
The primary cutaneous CD30+ lymphoproliferative disorders are linked by the common histological feature of large atypical lymphoid cells expressing CD30. 3 CD30 is a 120 kDa transmembrane cytokine receptor to the tumor necrosis factor receptor family and is preferentially expressed by activated lymphoid cells.
As well as the conditions discussed here, CD30 labels large proliferating T- and B-cell blasts in reactive lymphoid tissue,220. and 471. Reed–Sternberg cells and variants in classical Hodgkin lymphoma, proliferating cells of other B- and T-cell lymphomas, and other non-lymphoid neoplasms such as embryonal carcinoma. 471
The primary cutaneous CD30+ lymphoproliferative disorders include: primary cutaneous anaplastic large cell lymphoma (C-ALCL), lymphomatoid papulosis (LyP), and cases which do not fit clearly into these categories, borderline cases. Distinction between C-ALCL and LyP is not always possible on the basis of histological criteria and classification is based on clinical presentation and course as well as the histological appearance of lesions. Borderline cases are those that do not fit either classification; clinical follow-up may allow assignment to the appropriate group. 472 The Notch signaling pathway may play a role in the pathogenesis of this group of disorders. 473
Primary systemic anaplastic large cell lymphoma involves both lymph nodes and extranodal sites. The skin is commonly involved (21% of cases). Although skin lesions have identical morphology to those in C-ALCL, this form involves a different age group and has a different prognosis. Up to 85% of these tumors express anaplastic large cell lymphoma kinase protein (ALK). In most cases this results from the translocation t(2;5) in which part of the nucleophosmin (NPM) gene on chromosome 5 is juxtaposed to part of the ALK locus on chromosome 2 resulting in expression of the chimeric oncogenic tyrosine kinase NPM-ALK. The ALK-1 antibody is directed to the cytoplasmic portion of the ALK protein. 474 In 15–20% of cases expressing ALK there is a variant translocation and not the t(2;5) translocation. 475 It is discussed further later.
Primary cutaneous anaplastic large cell lymphoma
This form of lymphoma represented 9% of primary cutaneous lymphomas in the EORTC series. 3 This lymphoma occurs predominantly in older adults (median age of 60 years), and is rare in children and adolescents.476. and 477. There is a male preponderance. 478 Most patients have solitary or localized papules, nodules, and tumors which are red to violaceous in color and often ulcerated. Some single lesions resemble keratoacanthoma. 479 Single lesions may arise in uncommon sites such as the penis. 480 Partial or complete spontaneous regression occurs in up to 25% of cases. 481 This may be related to interactions of CD30 and its ligand (CD30L) and death receptor mediated apoptosis.482. and 483. Cases previously reported as regressing atypical histiocytosis represent this form of ALCL.484. and 485. A transient lesion has been reported associated with cyclosporine (ciclosporin) therapy. 486 Cases of C-ALCL demonstrate abnormal expression of the activator protein-1 (AP-1) transcription factor JUN. 487
The prognosis is very good but occasionally there are cases which disseminate in the skin and spread to involve lymph nodes and other sites, and these have a poorer prognosis.488.489. and 490. In one series, disease-specific survival for localized versus generalized disease was 91% and 50%, respectively. 489 Patients with multifocal disease are more likely to develop extracutaneous disease.488. and 491.
C-ALCL has been reported in association with atopic eczema, 492 psoriasis,493. and 494. internal malignancies,495. and 496. and B-cell lymphomas.497. and 498. It may also arise after solid organ transplantation.159.165.499.500.501.502. and 503. In very rare cases CD30+ circulating cells can be detected in the blood. 504
C-ALCL can arise in patients with previous LyP. In this situation it is difficult to distinguish it from histological type C LyP. It behaves like conventional C-ALCL in this situation and has a good prognosis. 505 It has been reported in patients with concurrent patch stage MF, 506 and in patients who develop LyP after C-ALCL. 507 LyP, MF, and C-ALCL appear to be clonally related in such patients.508. and 509.
Histopathology510
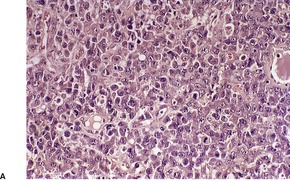
There is an infiltrate in the dermis and sometimes the subcutis which typically consists of confluent sheets of large, CD30+ cells (Fig. 41.8). The majority of these cells are typical anaplastic cells with round, oval, or indented nuclei (‘hallmark’ or ‘buttock’ cells), prominent nucleoli, and copious cytoplasm. In some cases the cells are more pleomorphic or immunoblastic in appearance. Some cells resemble Reed–Sternberg cells. A small cell variant with intermediate-sized atypical cells has also been described. 511 Cell type does not appear to affect prognosis. 488 There may be a significant component of small lymphocytes, eosinophils, neutrophils, and histiocytes which in some cases may obscure the neoplastic cells. 510 This is most prominent in the lymphohistiocytic and neutrophil-rich variants. 512 The epidermis is not infiltrated by tumor cells but there is often ulceration. Rare cases have a ‘signet-ring’-like morphology. 510 In some cases there is considerable epidermal hyperplasia. 479 Epidermotropism, tumor necrosis, vascular invasion and destruction, and nerve involvement are also present in some cases. 510 Xanthomatous change may be seen after radiation therapy. 513
 |
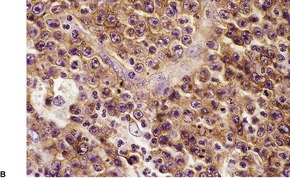 |
| Fig. 41.8 (A) Primary cutaneous anaplastic large cell lymphoma. (H & E) (B) Many cells exhibit a positive Golgi zone. (Immunoperoxidase stain for CD30) |
Immunohistochemistry and cytogenetics
More than 75% of the neoplastic cells are CD30+. 478 The majority are CD3+, CD4+, CD8−. A small number of cases are CD8+.15. and 514. Most but not all are EMA− and ALK−. 220,515 The presence of a positive reaction for ALK generally indicates cutaneous spread of primary systemic ALCL rather than C-ALCL. 516 Rare cases showing a positive cytoplasmic reaction for ALK without the usual t(2;5) translocation or variant translocations have been reported. 517 Positive reactions for CD15 in a cytoplasmic and Golgi zone pattern have been reported in a small number of cases. 505 Cells may express cytotoxic markers TIA-1, and granzyme B as well as CD56.518.519. and 520. Neoplastic cells may express the cutaneous lymphocyte antigen HECA 452 which is not usually expressed in primary systemic ALCL. 521 They also express MUM1. 310 In the vast majority of cases, the cells do not express EBV by PCR or in-situ hybridization.483.515. and 522.
The majority of cases are of T-cell type and exhibit clonal β or γ TCR gene rearrangements. 523 Atypical cells lack the t(2;5) translocation except in rare cases.524. and 525.
Clusterin expression, a marker for systemic anaplastic large cell lymphomas, can also be expressed by C-ALCL.526. and 527. Cells also express the cytokines CCR3 and CCR4,490. and 528. and the cell surface adhesion molecule CD44.529. and 530. CD95 (APO-1/Fas) is strongly expressed in C-ALCL and LyP and may play a role in regression of lesions. 531
Lymphomatoid papulosis
Lymphomatoid papulosis (LyP) is a chronic lymphoproliferative disorder characterized by the appearance of crops of papules, nodules, and sometimes large plaques at different stages of development. Lesions spontaneously regress after several weeks or months, sometimes resulting in atrophic scars. The clinical course may extend over decades. Initially, the lesions are smooth but later they become necrotic, crusted, and ulcerated.532.533.534.535.536.537. and 538. There may be only a few lesions, or many hundreds present during each exacerbation. 347 They occur mainly on the trunk and proximal parts of the limbs, but may also arise on the face, scalp, palms, and soles. Lesions rarely involve mucosal surfaces. 539 There is a predilection for females in the third and fourth decades of life. LyP also occurs in children.540.541. and 542. Rare clinical presentations include pustular lesions, 543 localized (agminated) lesions,544. and 545. lesions with a white halo, 546 and a pattern resembling hydroa vacciniforme. 547 It has been associated with the hypereosinophilic syndrome,548.549. and 550. atopic dermatitis treated with cyclosporine, 551 and parathyroid nodular hyperplasia. 552 Lesions in pregnant women may regress after delivery. 553 Keratoacanthoma has also been reported in association with LyP.554. and 555.
There is an increased frequency of prior, coexisting or subsequent lymphoproliferative disorders associated with LyP: this is most commonly mycosis fungoides or Hodgkin lymphoma.556.557.558.559.560. and 561. It has been claimed that in approximately 5–10% of cases, there is progression to a malignant lymphoma (Hodgkin lymphoma, MF, ALCL) or myeloproliferative disorder.347.373.556.562.563.564.565.566.567.568.569.570.571.572.573. and 574. Subsequent neoplasms may demonstrate identical clonal TCR gene rearrangements to those in LyP.561. and 575.
In a study of LyP associated with MF, LyP preceded MF in 67% of cases, followed MF in 19% of cases, and occurred concurrently in 14% of cases. 576
Clonal T cells similar to those in skin lesions have been rarely identified in bone marrow and blood.577. and 578.
LyP has an excellent prognosis. In a study of 70 patients by the EORTC group, no patient died of malignant lymphoma and the 5-year survival was 100%. Similar survival figures were reported in another series. 489
LyP is not associated with EBV579 or human herpesvirus type 6 and 7, but a recent study reports an association of LyP with human herpesvirus type 8 (HHV-8).515.580.581. and 582.
The histological appearance of LyP suggests it is a lymphomatous process and most would now regard this condition as a low-grade cutaneous T-cell lymphoma, part of a spectrum with C-ALCL. 583 Some still classify LyP as a ‘pseudolymphoma’. 584 Several studies have identified a clonal population of T cells in 60% of the lesions in LyP,535.585.586. and 587. but a clonal population is not always identified. 509 In a recent study, a clonal population was found in only 22% of archival cases of LyP by PCR. 523 Single cell analysis of CD30+ cells in one study found a single T-cell clone in all cases including anatomically and temporally separated lesions; CD30− background cells were polyclonal. 588
Because the histopathological and immunophenotypic features of LyP overlap with C-ALCL, the clinical appearance and course are important features in separating the two conditions and choosing treatment options. 3 Lesions of C-ALCL tend to be few and localized whereas LyP presents as recurring crops of multiple, widely dispersed lesions which involute spontaneously. Some cases have overlapping features and are classified as ‘borderline’. Spontaneous regression can occur in both LyP and C-ALCL. Borderline cases with follow-up may ultimately be classified as LyP or C-ALCL but overall have a favorable prognosis. 589 It has been suggested that in LyP, cell-mediated immune reactions involving the small lymphocytes may play a role in the spontaneous regression of lesions. 590
Histopathology591
The appearance of the lesions varies to a certain extent according to their age. There are three overlapping histopathological subtypes of LyP: types A, B, and C. 3
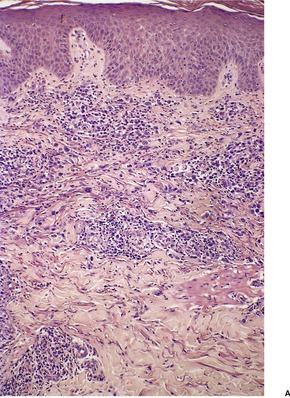
LyP type A is characterized by a wedge-shaped, mixed cellular dermal infiltrate which includes a variable number of large atypical cells similar to those in C-ALCL (Fig. 41.9). They may be multinucleate or resemble Reed–Sternberg cells. There is a background population of small lymphocytes, eosinophils, neutrophils, and histiocytes. Epidermotropism is variable but usually not prominent.
 |
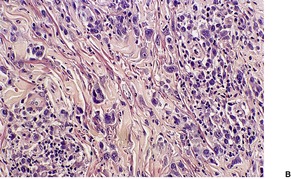 |
| Fig. 41.9 Lymphomatoid papulosis. (A) Type A histology. (B) There is a mixture of large atypical cells and small lymphocytes. (H & E) |
LyP type B is an uncommon pattern seen in 10% of cases. 15 It is characterized by a perivascular or band-like dermal infiltrate with epidermotropism. The predominant cell types are small to medium-sized lymphocytes with cerebriform nuclei. This picture resembles plaque stage MF and separation from MF may require clinical correlation. Large CD30+ cells seen in the other two forms are uncommon. Some lesions have overlap features of these two types. Different lesions in the same crop may show either of these patterns. 591
LyP type C lesions resemble C-ALCL and have a monotonous population of large atypical cells with relatively few admixed inflammatory cells.
Lesions in any of the subtypes may have focal spongiosis in the epidermis, parakeratosis with neutrophils, and ulceration. Epidermal hyperplasia is present in some cases. 592 Mitotic figures are common in all histological subtypes. There is some dermal fibrosis in resolving lesions, particularly in those that have ulcerated. There may be only a few atypical cells in late lesions. CD1a+ dendritic cells may be a conspicuous component of the cellular infiltrate. 593
A rare variant has been described in which the infiltrate is centered around hair follicles.594.595. and 596. Other variant patterns include a syringotropic pattern, LyP with epidermal vesicles, LyP with follicular mucinosis, 591 LyP with prominent myxoid change, 597 and LyP with angionecrosis. 598
Immunohistochemistry and cytogenetics
The large atypical cells in LyP types A and C are CD30+, CD3+, CD2+/−, CD4+/−, CD8− similar to the large cells in C-ALCL. Rare cases are CD8+.599. and 600. Cells are generally negative for CD15 and EMA but positive reactions have been reported in a few cases, the former having a cytoplasmic and Golgi zone pattern. 505 As in C-ALCL, the CD30+ cells may express cytotoxic markers TIA-1, perforin and granzyme B, and CD56;518.600.601.602. and 603. they are ALK−.518. and 601.
The atypical cells in the type B lesions are CD3+, CD4+, CD8−. Despite some reports, CD30+ cells may be present in this subtype and overlap cases with type A histology occur.15. and 591. The CD30+ cells may co-express CD134. 604
MUM1 (multiple myeloma oncogene 1) is expressed in the large cells in types A and C. 310 In one study, it was found that there was positive staining for MUM1 in 85% of cases of LyP but only in 20% of cases of primary C-ALCL. It was suggested that this was a useful marker is distinguishing between these two conditions. 605 Tumor necrosis factor receptor (TNFR)-associated factor 1 (TRAF1) has similarly been shown to be useful in distinguishing between LyP and C-ALCL, being expressed in 84% of the former and only in 7% of cases of anaplastic large cell lymphoma, primary and secondary. 606
SUBCUTANEOUS PANNICULITIS-LIKE T-CELL LYMPHOMA
The current WHO/EORTC classification confines this condition to those T-cell lymphomas which have a panniculitis-like histology and have an αβ+ T-cell phenotype. This group previously included lymphomas with a γδ phenotype. The latter group, which exhibits important differences in histology and behavior, is now classified separately as cutaneous γδ T-cell lymphoma (provisional) in primary cutaneous peripheral T-cell lymphoma, unspecified, of the WHO/EORTC classification. 15 Most of the earlier literature does not distinguish between these two groups.608.609.610. and 611. Other lymphomas can also involve the subcutis to varying degree. 611
Most if not all cases of so-called cytophagic histiocytic panniculitis (see p. 466) in the older literature, particularly the indolent forms, are not inflammatory but subcutaneous panniculitis-like T-cell lymphoma (SPTCL).608. and 609.
This condition presents as single or multiple nodules and plaques mainly on the legs but it may have a more general distribution. This may be associated with systemic symptoms such as fever and weight loss or a hemophagocytic syndrome. The lesions usually do not ulcerate. It rarely presents as alopecia. 612 Typically there is an indolent course without extracutaneous spread.613.614.615. and 616. Cases complicated by a hemophagocytic syndrome have a more aggressive course.613. and 617. When the condition is carefully defined, the 5-year survival is in the region of 80%.611. and 618.
Histopathology15.615. and 618.
The lymphomatous infiltrate is confined to the subcutis where it predominantly involves lobules to produce a lobular panniculitis-like pattern (Fig. 41.10). 618 The atypical lymphocytes comprising the infiltrate are small, medium, or large with irregular hyperchromatic nuclei. Numerous histiocytes are also present. The atypical lymphocytes characteristically rim adipocytes to form a lace-like pattern, but this is not entirely specific.611. and 618. Apoptotic bodies (karyorrhectic nuclear fragments) are invariably present. A component of large phagocytic histiocytes (‘bean-bag histiocytes’) containing cell debris or red blood cells (hematophagocytosis) is sometimes seen. Fat necrosis with foamy macrophages may also be present. In early stages of the disease the cellular infiltrate may be less atypical. 616 Plasma cells are present in some cases but neutrophils and eosinophils are generally absent. Angiocentricity, angioinvasion, and large areas of necrosis are not typical of this entity.
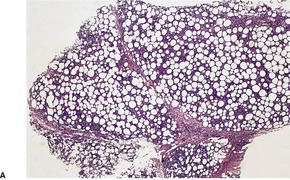 |
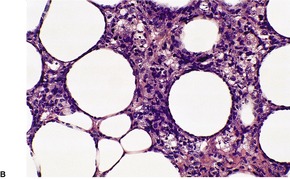 |
| Fig. 41.10 (A) Subcutaneous panniculitis-like T-cell lymphoma. (B) There are atypical lymphoid cells ringing adipocytes and histiocytes with cell debris. (H & E) |
Immunohistochemistry and cytogenetics
The neoplastic cells by definition have a TCR α/β, T-cell phenotype and are CD3+, CD4–, and CD8+. They also express cytotoxic markers TIA1 and granzyme B. CD30 and CD56 are not expressed. 15 They do not show evidence of EBV infection.
Recent genomic studies have shown that SPTCL is a uniform entity with characteristic gains of 5q and 13q as well as other abnormalities observed in cutaneous T-cell lymphoma types. 619
EXTRANODAL NK/T-CELL LYMPHOMA, NASAL TYPE
NK and cytotoxic T cells are closely related in ontogeny and function and they have overlapping immunophenotypic features. 620
Extranodal NK/T-cell lymphoma, nasal type (E-NK/NT) is the best characterized of the NK/T group of lymphomas.621.622. and 623. The presence of EBV in almost all cases suggests a pathogenic role for this virus in the generation of this lymphoma. Rarely, there is documentation of preceding chronic active or continuous EBV infection.624. and 625. This form of lymphoma is common in Asia, South and Central America and Mexico, but is uncommon in the United States and Europe. It is almost always associated with EBV when carefully defined, even in white people. 626 Males are affected predominantly, with a median age in the fifth decade. 627 Children and adolescents are rarely affected. 628
It presents most commonly in the nasal region, often with midline facial destructive disease (‘lethal midline granuloma’) or a nasal mass. 629 The extranasal form of this lymphoma presents most commonly in the skin and subcutis and less commonly in the upper respiratory tract, gastrointestinal tract, testis, and spleen. 627 These organs are also the sites to which the nasal form most commonly disseminates. 630 Nasal metastasis may follow skin presentation. 631 Unlike lymphomatoid granulomatosis, the lung is rarely involved. Nodal involvement is also very uncommon. 632
Cutaneous lesions include ulcerated tumors, subcutaneous nodules resembling a panniculitis, erythematous plaques, purpura, bullous lesions, and a macular papular rash. 622 Lesions occur on the trunk and extremities. 633
A hemophagocytic syndrome is sometimes a complication. It may arise in a background of immunosuppression. 634
This is an aggressive lymphoma with a short median survival and high mortality rate.626. and 635. In one series the median survival was 5 months. Survival was slightly better when confined to the skin (median survival 27 months).636. and 637. Some cases have overlap features with aggressive NK/T-cell leukemia. 626
An extremely rare intravascular variant has been described. 638
Histopathology639
There is a diffuse or angiocentric and periappendageal cellular infiltrate which involves the dermis and subcutis (Fig. 41.11). The infiltrate consists of variably sized small and intermediate-sized lymphocytes. Large cells may predominate in some cases. 639
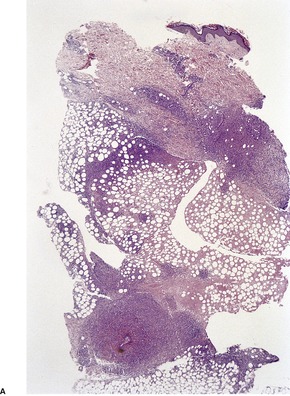 |
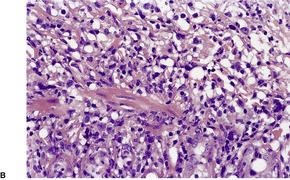 |
| Fig. 41.11 Extranodal NK/T-cell lymphoma. (A) An angiocentric pattern of infiltration is evident. (B) A polymorphous lymphoid infiltrate is present in the wall of a vessel. (H & E) |
Angiocentricity is associated with angioinvasion of vessel walls, sometimes with fibrinoid necrosis. 623 Although this is common in this lymphoma it is not always present. 623 Furthermore, it is not a specific feature and can be seen in other cutaneous lymphomas including B-cell neoplasms. 640
Cellular pleomorphism may be slight and this feature, together with a background population of other inflammatory cells including eosinophils and plasma cells, may lead to a misdiagnosis of an inflammatory reaction such as a lobular panniculitis.625.640. and 641. Involvement of the subcutis can mimic panniculitis-like T-cell lymphoma, particularly if angiocentricity is absent. Phagocytic macrophages and ‘rimming’ of adipocytes may be present. 641
Zonal necrosis and karyorrhexis are frequently seen. This may be due to release of cytotoxic granules and the induction of apoptosis,642. and 643. or the production of TNF-α (tumor necrosis factor-α).
Epidermotropism is seen in some cases. 635
Immunohistochemistry and cytogenetics
The tumor lymphocytes are CD56+, CD2+, and surface CD3−. They express cytoplasmic CD3ε and the cytotoxic proteins TIA-1, perforin, and granzyme B. CD2, CD7, and CD8 are occasionally expressed.623. and 644. Small numbers of CD30+ cells are seen in some cases.625. and 645. EBV can be detected in most cases by in-situ hybridization for EBER (EBV-encoded small RNA) and occasionally by immunohistochemistry (LMP-1).
There is no clonal rearrangement of the TCR (germline) in almost all cases. There can be clonal TCR gene rearrangement in rare cases with a cytotoxic T-cell phenotype.15. and 646. A related CD8+, CD30+, CD56+, CD3ε+, EBV+ aggressive neoplasm with clonal TCR gene rearrangement has been described.
A variety of cytogenetic abnormalities have been reported; deletions of 6q are the most common. 639
Hydroa vacciniforme-like lymphoma
This is a lymphoproliferative disorder of NK cells and rarely T cells which has overlap features with NK/T-cell lymphoma, nasal type, and is considered by some a variant of that condition. 639 Clinically it mimics hydroa vacciniforme (see p. 536), an eruption related to sun exposure in which there are edema, vesicles, necrosis, and scars on the face and dorsa of the limbs.647. and 648. It is seen mainly in children from Asia or Central and South America but it may also occur in adults. 649
Clinically there is a papulovesicular eruption on sun-exposed skin, particularly the face. The disease may be precipitated by insect bites. There may be associated systemic symptoms of fever and weight loss; lymphadenopathy and hepatosplenomegaly may be present. A hemophagocytic syndrome may occur.
The prognosis is said to be poor, but in some cases at least it appears to be better than in typical E-NK/NT.639. and 650.
PRIMARY CUTANEOUS PERIPHERAL T-CELL LYMPHOMA, UNSPECIFIED
This heterogeneous group of cutaneous T-cell lymphomas does not fit into one of the well-defined subtypes of T-cell lymphoma.
Primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma
When carefully defined, primary cutaneous CD8+ epidermotropic cytotoxic T-cell lymphoma appears to be a clinicopathological entity distinct from classical MF, pagetoid reticulosis, and other cutaneous lymphomas which may have a CD8+ phenotype.367.652. and 653. It is characterized clinically by generalized patches, plaques, papulonodules, and tumors mimicking disseminated pagetoid reticulosis. There is systemic spread to the blood, lungs, testis, central nervous system, and oral cavity but not usually to lymph nodes. 654 It has an aggressive clinical course with a median survival of 32 months.41.367. and 654.
Histopathology
In early lesions there is an epidermal infiltrate of atypical lymphocytes in a pagetoid pattern. Fully developed lesions are characterized by a band-like dermal infiltrate of atypical cells as well as by diffuse infiltration of the epidermis. Atypical cells vary from small/medium to large pleomorphic cells and immunoblasts. The epidermis is acanthotic and there are frequently secondary changes of spongiosis, vesiculation, and necrosis. Unlike mycosis fungoides, a diffuse epidermal infiltrate is present in the tumor stage. Sweat gland and hair follicles are frequently involved, sometimes producing lymphoepithelial-like lesions. Rare eosinophils and plasma cells are present.
Immunohistochemistry and cytogenetics
The neoplastic lymphocytes have a CD3+, CD8+, CD7+, CD45RA+ phenotype and also express the cytotoxic markers TIA-1, granzyme B, and perforin. There is a high Ki-67 proliferation index. Unlike pagetoid reticulosis, these cases do not express HECA 452. Clonal rearrangement of the TCRγ gene has been demonstrated in all cases.
Cutaneous γ/δ T-cell lymphoma
This cutaneous lymphoma was previously regarded as a variant of subcutaneous panniculitis-like T-cell lymphoma (SPTCL) but has been separated from this group because of significant clinical and phenotypic differences. 15 This form of cutaneous lymphoma presents as plaques, necrotic nodules and tumors, particularly on the extremities. There may be involvement of mucosal and other extracutaneous sites. 655 A hemophagocytic syndrome may be present as in SPTCL. 617 Unlike that form of lymphoma, there is usually an aggressive clinical course. In one series the median survival was 15 months. 656 Those patients with involvement of the subcutis have a worse prognosis than those with more superficial disease.
Histopathology
There is an infiltrate of medium to large atypical lymphocytes which may involve the epidermis, dermis, or subcutis sometimes with angiotropism and necrosis.616. and 657. Ulceration is sometimes present. Involvement of the subcutis resembles the pattern seen in SPTCL with ‘rimming’ of adipocytes. Apoptotic bodies are commonly observed.
Immunohistochemistry and cytogenetics
This lymphoma is a monoclonal proliferation of T lymphocytes that express the γδ phenotype. This can be demonstrated by PCR on paraffin sections. Staining for TCRδ (TCRδ1) by immunoperoxidase techniques can only be carried out on frozen section material. 656 The cells are negative for βF1 in paraffin sections, a marker for the αβ phenotype. The atypical lymphocytes are CD3+, CD2+, CD56+, and also express cytotoxic markers such as TIA1 and granzyme B. Most are CD4− and CD8− but occasional cases are CD8+. 656 They are generally negative for EBV but rare EBV+ cases have been reported from Asia.617. and 658.
Primary cutaneous CD4+ small/medium pleomorphic T-cell lymphoma
This rare lymphoma is defined by the presence of a cutaneous infiltrate of small to medium-sized T-lymphocytes which are CD4+, without pre-existing early lesions typical of mycosis fungoides. Cases with a CD8+ phenotype have also been reported659 but have been excluded from this category in the WHO/EORTC classification. 15 The usual presentation is as a solitary plaque or tumor on the face, neck, or upper trunk but less commonly there may be multiple lesions present.660.661. and 662. The prognosis is usually excellent, particularly when there is a single nodule or localized skin involvement.3.660.662. and 663.
Histopathology
There is a nodular or diffuse infiltrate in the dermis which may extend into the subcutis, and rarely into the epidermis focally. 15 The infiltrate is polymorphous and may mimic benign lymphoid hyperplasia. The predominant cells are small to medium pleomorphic T cells admixed with reactive B cells, plasma cells, and histiocytes. 660 Granulomas may be seen in the infiltrate. 269
Immunohistochemistry and cytogenetics
The atypical T cells have a CD3+, CD4+, CD8−, CD30− phenotype and there may be loss of CD7 and CD2. There is clonal rearrangement of TCR genes. This latter feature is useful in distinguishing this lymphoma from benign simulants or cutaneous B-cell lymphomas.
CUTANEOUS B-CELL LYMPHOMAS
Cutaneous follicle center cell lymphoma and extranodal marginal zone B-cell lymphoma of MALT type represent 90% of all forms of cutaneous B-cell lymphoma. They share similar clinical presentations, response to therapy, and an excellent prognosis. 664
MARGINAL ZONE B-CELL LYMPHOMA
The majority of low-grade B-cell lymphomas appear to be of this type.665.666. and 667. The rubric primary cutaneous marginal zone B-cell lymphoma (PCMZL) also encompassed those lesions previously referred to as cutaneous immunocytoma668. and 669. and cutaneous follicular hyperplasia with monotypic plasma cells.670 The current WHO/EORTC classification includes cutaneous plasmacytoma without underlying myeloma in this category because of overlap between this condition and plasma cell-rich lymphomas in this group (see p. 998). 15 European and British cases, but not those from the United States or Asia, may be associated with Borrelia burgdorferi infection.671.672.673.674.675. and 676.
This form of cutaneous lymphoma occurs most frequently in adults with a male predominance.676.677. and 678. In one large series the median age was 50 years but there is a wide age range. 677
Lesions occur predominantly on the arms and trunk677 but many other sites such as the head and neck may also be involved.667.668.676.678.679.680.681. and 682. They are frequently multifocal in contrast to primary cutaneous follicle center cell lymphoma. A dermatomal distribution has been described. 683 This form of lymphoma can also involve the lip, tongue, and oral mucosa. 684 The lesions are red to purple papules, nodules, and plaques. Lesions have a tendency to recur locally or at other skin sites but dissemination to extracutaneous sites is very rare. These include lymph nodes and other sites, particularly other organs where MALT-type lymphomas occur.667. and 685. The lesions may spontaneously regress, sometimes leading to secondary anetoderma. 686 Cases with a single lesion or localized cluster of lesions are more likely to have a sustained complete remission than those with multiple non-localized lesions. 687 The long-term prognosis is excellent with a 5-year survival reported at 98%. 688 Large cell transformation has been reported and is associated with a worse prognosis.39. and 689. Rarely systemic spread leads to death.671.678. and 689. PCMZL has been reported with chronic lymphocytic leukemia, 690 at the site of vaccination, 691 and following fluoxetine therapy. 692
Histopathology38.56.665. and 693.
There is a dermal cellular infiltrate that may extend into the subcutis but spares the epidermis. The infiltrate has a nodular or diffuse pattern characterized by reactive lymphoid follicles surrounded by paler zones containing a mixed population of cells that includes small lymphocytes, centrocyte-like cells with cleaved nuclei, monocytoid B cells with round nuclei and more prominent pale cytoplasm, lymphoplasmacytoid cells, 694 and plasma cells. Where there is a more diffuse pattern, the mantles of follicles may be obliterated by the neoplastic component (follicular colonization). 678 In the interfollicular areas there are occasional large blasts as well as variable numbers of histiocytes and eosinophils. Plasma cells are often present at the periphery of the nodules or beneath the epidermis. Unlike the other extranodal marginal zone B-cell lymphomas (MALT lymphomas), lymphoepithelial lesions involving the skin appendages are rare but eccrine ducts or pilosebaceous units may be infiltrated by lymphoid cells.664.678. and 695. In cases previously reported as immunocytoma there are often more diffuse infiltrates with few reactive follicles and more prominent lymphoplasmacytoid cells (lymphoid nucleus and plasma cell-like cytoplasm), and plasma cells (Fig. 41.12). Dutcher bodies (intranuclear PAS-positive inclusions) may be present in these cells and are considered to be indicative of a neoplastic process. Lesions that consist overwhelmingly of monotypic plasma cells that have been called cutaneous plasmacytoma (see p. 943) are also probably variants of cutaneous marginal zone B-cell lymphoma. 696
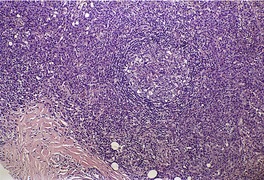 |
| Fig. 41.12 Marginal zone lymphoma of MALT type. A reactive follicle is present with sheets of monotypic plasma cells. (H & E) |
Immunohistochemistry and cytogenetics
All variants of the small neoplastic cells express CD20 and CD79a. The latter is also expressed by plasma cells which also express CD38 and CD138. The lymphocytes also express bcl-2 but not CD5, CD10, CD43, cyclin D1, or bcl-6. 681 Plasma cells and lymphoplasmacytoid cells show cytoplasmic immunoglobulin light chain restriction. The scattered large blastic cells can be CD30+. Aberrant nuclear bcl-10 expression may be observed.676. and 697.
Immunoglobulin heavy chain (IgH) genes are clonally rearranged. Rare cases have shown a dual B- and T-cell phenotype. 698 A proportion of cases exhibit trisomy 3, 699 the t(14;18)(9q32;q21) translocation or the t(3;14)(p14.1;q32) translocation which involve the immunoglobulin heavy chain locus (IGH) and MALT1 gene and the FOXP1 gene, respectively.700. and 701. The translocation involving IGH and MALT1 is reported to be uncommon, however. 702 The t(14;18)(q32;q21) IGH/BCL2 translocation has also recently been reported in PCMZL. 703 Frequent inactivation of p15 and p16 genes has been observed, most commonly as a result of promoter hypermethylation. 704
FOLLICLE CENTER CELL LYMPHOMA
It has only been comparatively recently that this low-grade primary cutaneous lymphoma has been established as an entity that is distinct from conventional nodal follicular lymphoma,38.490.680. and 705. and primary cutaneous marginal zone lymphoma.
Lesions that historically were called reticulohistiocytoma of the dorsum, Crosti’s lymphoma, and large cell lymphocytoma are examples of this form of lymphoma.706.707.708. and 709. This is a lymphoma of adults; pediatric cases are extremely rare. 710 It has been reported in a radiotherapy field. 711 Most lesions arise on the head and neck, and trunk and are usually single, erythematous plaques or tumors. When on the trunk, there may be plaques or tumors surrounded by smaller papules. Multiple separate lesions do occur but are uncommon. The lesions can increase in size with time and recurrences are common but extracutaneous spread is rare.710. and 712. The prognosis is excellent with a 5-year survival of 95%.713. and 714. The prognosis does not depend on whether the growth pattern is follicular or diffuse, or whether there is localized or multifocal skin disease. 714 There may be a worse prognosis in those with a diffuse large cell histology and strong expression of bcl-2. 715
Histopathology
There is a nodular or diffuse infiltrate involving the dermis and sometimes the subcutis. The infiltrate may have a vertical orientation that is centered on skin appendages and neurovascular bundles. 716 The infiltrate may rarely involve the subcutis only. 717 There may be a follicular, follicular and diffuse, or a diffuse pattern. The last is distinct from other forms of large cell lymphoma such as diffuse large cell lymphoma, leg type. A prominent follicular pattern is most typically seen in lesions from the head and neck (Fig. 41.13). Follicles are often variable in size and may fuse together. 38 Unlike reactive follicles, there is usually an absent or poorly developed mantle or fragmentation of the germinal center with invagination of the mantle zone resembling nodal progressive transformation of germinal centers. This has been referred to by some as a ‘floral pattern’. 716 The follicle centers contain varying proportions of large and small centrocytes with cleaved nuclei and centroblasts with non-cleaved nuclei and prominent nucleoli. The distribution of these cells lacks the polarization into predominantly centroblastic of centrocytic zones seen in reactive germinal centers. Tingible body macrophages are absent or few in number (Fig. 41.14). Occasionally the follicle center cells have a spindled appearance. There are surrounding infiltrates of small lymphocytes sometimes with histiocytes, eosinophils, and plasma cells. The diffuse forms have sheets of large follicle center cells including large centrocytes, multilobated cells, and variable numbers of centroblasts and immunoblasts. Unlike other diffuse large cell lymphomas the infiltrate does not consist of a monotonous population of centroblasts and immunoblasts. 714
 |
| Fig. 41.13 Follicle center cell lymphoma. The follicular structures may be large and irregular. (H & E) |
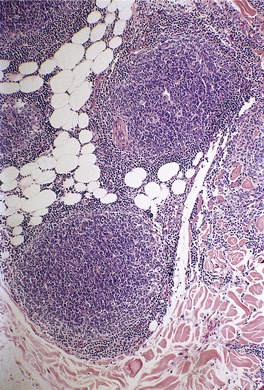 |
| Fig. 41.14 Follicle center cell lymphoma. There is a poorly formed mantle zone and no tingible body macrophages. (H & E) |
The small number of spindle-cell B-cell lymphomas reported in the literature probably represent variants of diffuse follicle center cell lymphoma as the tumor cells mostly resemble elongated and distorted large centrocytes.718.719. and 720. There may be associated myxoid change in the stroma. 721
Immunohistochemistry and cytogenetics
The follicular pattern is emphasized by CD21 staining of follicular dendritic cells. Only ragged remnants of these cells are present as the pattern of infiltration becomes more diffuse. The neoplastic cells are CD10+, CD20+, CD43+, CD79a+, and bcl-6+. CD10 and bcl-6 also demonstrate small groups of cells outside the germinal centers. Reactive T cells are present in follicles and interfollicular areas. MIB-1 usually reveals a low proliferation fraction in the follicle centers. Plasma cells are polyclonal. There is variable staining of the follicle center cells for bcl-2 but it is absent in most cases and if present may be only in a small proportion of cells, unlike follicular lymphoma of lymph nodes.38.679.680.722.723.724.725. and 726. A strong positive reaction raises the possibility of a secondary follicular lymphoma. A small number of cases are positive for MUM1. 727
Clonal rearrangement of IgH can be demonstrated in over 50% of cases. 728 Unlike follicular lymphoma of lymph nodes the t(14;18) translocation is not found in most cases.729. and 730. A recent study has shown that in 41% of cases the translocation t(14;18) was demonstrated by FISH techniques but in no cases by PCR. 728 Mutations of the BCL-6 gene have been detected.723. and 728.
Differential diagnosis731
The main differential diagnosis for both primary cutaneous marginal zone B-cell lymphoma (CMZL) and cutaneous follicle center cell lymphoma (FCL) is cutaneous lymphoid hyperplasia (LH). All have a follicular growth pattern in part. Separation of these conditions still remains difficult but can be achieved in most cases by a combination of histopathology, immunohistochemistry, and B-cell gene rearrangement studies.
Some histological features previously considered to be helpful in separating lymphoma from lymphoid hyperplasia are now known to be unreliable, e.g. ‘bottom heavy’ versus ‘top heavy’ cellular infiltrates. 716 Although malignant infiltrates are more likely to be ‘bottom heavy’ and deep, often involving the subcutis, this pattern can also be seen in benign infiltrates.
In florid reactive lymphoid hyperplasia, the lymphoid follicles may have a poorly formed or absent mantle zone. Follicle centers in LH and CMZL tend to show zonation, have obvious tingible body macrophages, and more centroblasts and immunoblasts. Follicle centers in FCL lack zonation, have a predominance of large cleaved centrocytes and no, or only rare tingible body macrophages.
A useful immunohistochemical feature for distinguishing FCL from LH is the presence of small clusters of CD10+/bcl-6+ cells in the interfollicular zones in the former. There is a low MIB-1 proliferation fraction in FCL compared with LH. The follicle centers are predominantly negative for bcl-2 in all three conditions. Rare cases of FCL are positive for bcl-2 but must be distinguished from secondary nodal follicular lymphoma. 731
The margins of the follicle centers, delineated by follicular dendritic cells demonstrated by CD21, tend to be well defined in LH and CMZL and more irregular in FCL. Plasma cells in CMZL are monoclonal, and polyclonal in FCL and LH.
The presence or otherwise of other inflammatory cells such as histiocytes, plasma cells, and eosinophils is not helpful in separating these conditions.
DIFFUSE LARGE B-CELL LYMPHOMA
In the general WHO classification of lymphomas, diffuse large cell lymphomas are composed of large cells with nuclei at least twice the size of a small lymphocyte and usually larger than a macrophage nucleus (Fig. 41.15). Cell types include centroblasts or large non-cleaved cells (cells with scant basophilic cytoplasm and round nuclei with multiple medium-sized, usually peripheral nucleoli), and immunoblasts (large cells with more conspicuous, often amphophilic cytoplasm, oval nuclei and a large single, central nucleolus). Other cell types include large centrocytes (large cleaved cells with small nucleoli), multilobated cells, and anaplastic large cells identical to T-cell and null-cell anaplastic lymphoma. 1 Nodal diffuse large B-cell lymphoma may present in the skin.732.733. and 734.
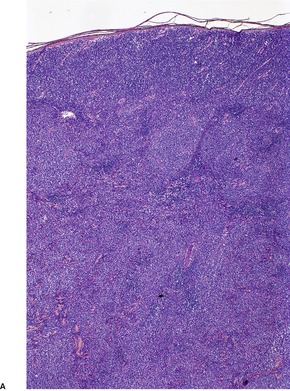 |
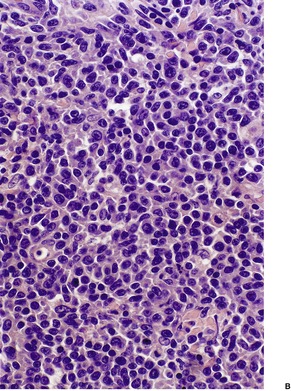 |
| Fig. 41.15 (A) Diffuse large B-cell lymphoma. (B) The large B cells are predominantly centroblasts and immunoblasts. (H & E) |
Vermeer et al, and subsequently the authors of the EORTC classification, distinguished a form of diffuse large cell lymphoma which occurred predominantly on the leg – large B-cell lymphoma of the legs. 735 This appears to be a distinct type of primary lymphoma that subsequently has been shown to occur at sites other than the leg.15. and 736. There are clinical, morphological, and immunohistological features which distinguish this lymphoma from the diffuse form of primary follicle center cell lymphoma which has histological features of a large cell lymphoma.
The concept of primary cutaneous diffuse large B-cell lymphoma, leg type has been controversial but the current WHO/EORTC classification now distinguishes this lymphoma as a specific subtype of diffuse large B-cell lymphoma. The remaining primary large B-cell lymphomas are classified under the rubric primary cutaneous diffuse large B-cell lymphoma, other. This group includes other rare cases of large B-cell lymphoma occurring in the skin including intravascular large B-cell lymphoma, T-cell rich/histiocyte-rich B-cell lymphoma, and anaplastic or plasmablastic types.
Primary follicle center cell lymphoma with a diffuse large cell morphology is classified with that subtype.
In a recent study of cutaneous large B-cell lymphomas, 90.3% of cases were able to be classified as either primary cutaneous diffuse large B-cell lymphoma, leg type or primary cutaneous follicle center cell lymphoma. 727 Both these lymphomas appear to be of germinal center origin.737. and 738. Gene expression profiling supports the subdivision of the two main types of cutaneous large cell lymphoma.27. and 739.
In both these groups, expression of bcl-2, OCT-2, and MUM1 are associated with a poor prognosis and staining with bcl-6 with a good prognosis.740. and 741. Location of primary cutaneous large cell lymphoma on the leg was an independent prognostic factor in some series. 742
Genetic aberrations including numerical alterations were identified in 76% of cutaneous large B-cell lymphoma regardless of the type. 743
DIFFUSE LARGE B-CELL LYMPHOMA, LEG TYPE
Although this lymphoma occurs most frequently on the legs (72% in one series), 744 tumors with identical features can occur at other sites. It is a tumor of the elderly, mean age 76 years, with a female predominance of cases. 744 It has been reported in a background of chronic lymphedema of the leg or chronic venous insufficiency.727.745. and 746. The classical presentation is of one or more rapidly growing erythematous tumors on one or both lower legs; ulceration is common. Occasional lesions are annular. 747 The lesions relapse frequently after therapy, and extracutaneous spread is common. In a recent study the overall 5-year disease-specific survival was 41%. In this series the central nervous system was the most frequent site of visceral dissemination. 744
Relapse as an intravascular large B-cell lymphoma has been reported. 748
Histopathology
There is a diffuse infiltrate in the dermis and frequently the subcutis of large lymphocytes with the features of centroblasts and immunoblasts. Mitotic figures are common. Other cellular elements are uncommon. The cells usually have round nuclei and lack the cleaved appearance of the predominant large lymphocytes seen in primary follicle center cell lymphoma.
Immunohistochemistry and cytogenetics
The cells are CD20+, CD79a+, and bcl-6+. Most express bcl-2 and MUM1/IRF4744 unlike cutaneous follicle center cell lymphoma. Some cases also express FOX-P1 (a member of the FOX-P transcription factors). 727 A minority of cases express CD10. 727 Rare cases are CD30+.727. and 749.
Treatment of B-cell lymphomas including large B-cell lymphoma, leg type with rituximab (an anti-CD20 monoclonal antibody) therapy may result in recurrences failing to stain for CD20, a potential pitfall for dematopathologists.751.752.753.754.755.756.757. and 758. In this situation staining for Pax5, a pan B cell marker, may be useful. 759
DIFFUSE LARGE B-CELL LYMPHOMA, OTHER
The best defined lymphoma in this group is intravascular large B-cell lymphoma and it is given a separate designation in the WHO/EORTC classification. Other diffuse large cell lymphomas include: anaplastic and plasmablastic subtypes, T-cell/histiocyte rich large B-cell lymphomas, and other less well-defined entities.
Intravascular large B-cell lymphoma
Intravascular large B-cell lymphoma (IVLBL) is a rare form of lymphoma which commonly presents with skin lesions or pain. It has been termed ‘intravascular lymphomatosis’ and ‘angiotropic large cell lymphoma’.763. and 764. It is characterized as an intravascular proliferation of large atypical lymphoid cells. In the majority of cases they are B cells, but rare cases with a similar histological appearance have a T- or NK-cell phenotype.765.766.767. and 768. It has been suggested that the lack of expression of CD29 (β1 integrin) and CD54 (ICAM-1) adhesion molecules by the lymphoma cells is responsible for the disseminated intravascular pattern, these molecules being important in lymphocyte trafficking and transvascular migration. 769
The median age at presentation is 70 years and there is an approximately equal sex incidence. 770
Cutaneous lesions are the dominant presenting feature in a third or more of cases. 770 They take the form of erythematous to blue macules, plaques and nodules,771.772.773. and 774. and generalized telangiectasia. 775 In some cases there is only very subtle figurate erythema. Skin lesions may mimic panniculitis such as erythema nodosum, 776 hemangiomas, 777 angiolipomas, 778 Kaposi’s sarcoma, 779 thrombophlebitis, or erysipelas. 773
Lesions occur predominantly on the upper and lower limbs, and trunk. 761 Pain and ankle edema may accompany the skin lesions.
A subgroup of patients representing about 26% of cases, and who are invariably female, present with cutaneous manifestations only. 770 Approximately a third of cases present with neurological symptoms770 including a variety of motor and sensory deficits and dementia.770. and 780. Other presentations include disseminated intravascular coagulation, 780 and, almost exclusively in Asian countries, a hemophagocytic syndrome.781. and 782. Patients in this group rarely exhibit involvement of the skin or central nervous system. There is commonly an elevation of lactic dehydrogenase serum levels and anemia. 783 Hepatosplenic and bone marrow involvement is common but nodal involvement is rare. 770
There may be an associated extravascular large cell lymphoma, either nodal or extranodal.784. and 785. There are also reports of an association with follicular lymphoma and MALT lymphoma.769. and 786.
The prognosis is poor: 85% have a fatal outcome. The prognosis is better in those with disease restricted to skin; 10% have a fatal outcome in this group.773.787. and 788.
Histopathology
Blood vessels in the dermis and subcutis are partially or completely occluded by large atypical lymphoid cells (Fig. 41.16). The cells are several times larger than endothelial cells and have a high nuclear:cytoplasmic ratio. Nuclei are round to oval and nucleoli are often prominent. Mitotic figure, including atypical forms, are frequently seen. Fibrin thrombi are often present in vessels, either with or without atypical cells. Extravascular neoplastic cells may also be found in up to 20% of cases. 3 Occasionally associated perivascular inflammatory changes may mask the intravascular lymphoma. 789
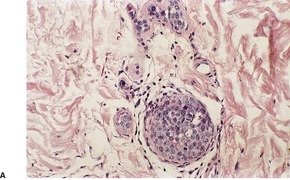 |
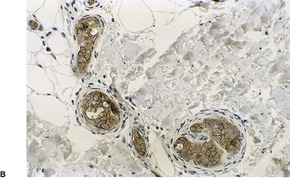 |
| Fig. 41.16 (A) Intravascular large B-cell lymphoma. (H & E) (B) The intravascular cells are positive for CD20. (Immunoperoxidase stain for CD20) |
The upper dermis may be spared and a superficial biopsy may not show the diagnostic changes. 787 On the other hand, a ‘blind’ biopsy of apparently normal skin may show diagnostic changes.785.790. and 791.
Immunohistochemistry and cytogenetics
The atypical lymphocytes are CD20+, CD79a+, and CD3−. There is variable staining for CD5 (38%), CD10 (13%), bcl-6 (26%), MUM1 (95%), and bcl-2 (91%). 792
Plasmablastic lymphoma
Plasmablastic lymphoma is a rare B-cell lymphoma most often associated with HIV-AIDS or other forms of immunosuppression such as organ transplantation.795. and 796. It may take an oral form or be associated with multicentric Castleman’s disease (see p. 946). 797 It is associated with both EBV and HHV-8 infection.
Skin involvement alone is uncommon. In the skin, there are solitary or multiple, purple-red nodules or tumors which may ulcerate.798. and 799. The extremities are most commonly involved. 800 Apart from the oral cavity, other common sites of involvement include the bone, soft tissues, and gastrointestinal tract.
It has an aggressive clinical course with a median survival of 7 months in one series. 801
Histopathology801
Immunohistochemistry and cytogenetics
The tumor cells are CD45+, CD38+, CD138+, CD20−, CD3–, CD5−, CD30−, bcl-6−, and Pax5−. There may also be monotypic cytoplasmic staining for immunoglobulin light chains.
The tumor is strongly associated with EBV and the cells are positive for EBER. The cells may also be positive for HHV-8 by immunohistochemistry.
T-cell/histiocyte-rich B-cell lymphoma
Primary cutaneous T-cell rich/histiocyte-rich B-cell lymphoma is a very rare variant of large B-cell lymphoma which is characterized by the presence of small numbers of neoplastic large B cells surrounded by large numbers of reactive small T cells and histiocytes.802.803. and 804. The relative proportions of large B cells and reactive T cells have not been clearly defined. 805 The nodal form of this lymphoma rarely involves the skin secondarily.806. and 807. The average age of patients in several series was 42 years. The lesions have no particular anatomical distribution. 808 It may be associated with HIV infection. 805 An angiocentric form has been described which has similarities to lymphomatoid granulomatosis (see below). 808 The clinical course is variable in the small number of cases described; some cases have progressed to disseminated disease.
Histopathology
Diagnosis is difficult because of the polymorphous nature of the infiltrate and the small number of neoplastic cells. There are scattered large B cells which may resemble centroblasts, immunoblasts, or Reed–Sternberg-like cells. 803 The majority of cells are small lymphoid cells with variable numbers of histiocytes, eosinophils, and plasma cells. The differential diagnosis includes Hodgkin lymphoma, peripheral T-cell lymphoma, and benign lymphoid hyperplasia.
Immunohistochemistry and cytogenetics
Diagnosis requires demonstration of a monoclonal population of B cells by PCR for IgH rearrangements or other techniques. The neoplastic B cells are CD20+, CD79a+, and positive for other pan B-cell markers. Some are bcl-2+. 809 The background lymphocytes are predominantly small T cells which are CD3+.
Lymphomatoid granulomatosis
The concept of lymphomatoid granulomatosis (LG) has evolved since it was first described by Liebow et al in 1972. 810 It was originally defined as an angiocentric, angiodestructive, lymphohistiocytic disorder which affected the lungs predominantly, but had frequent extrapulmonary manifestations. It was for some time considered to be a T-cell proliferative disorder because of the large number of T cells in the lesions, although most studies failed to demonstrate a clonal proliferation of T cells. 811
Epstein–Barr virus (EBV) was identified in most pulmonary cases and some cutaneous lesions, implicating it in the pathogenesis of LG.812. and 813. Subsequent studies have localized EBV to a population of large atypical cells which may only represent a minor component in the polymorphous infiltrate.814.815. and 816. PCR studies have subsequently demonstrated a monoclonal B-cell proliferation.
Lymphomatoid granulomatosis is now regarded as an EBV-driven disorder of B cells with accompanying reactive T cells and histiocytes.816. and 817. There is a spectrum of histological grade818 and clinical behavior which reflects the proportion of large EBV-infected neoplastic B cells. Grade 3 lesions, in which the large B-cell proliferation is obvious, are regarded as diffuse large B-cell lymphoma. 2 Chemokines have been implicated in the vascular damage. 819
Skin lesion are found in 40–60% of cases; they may be the presenting complaint820.821.822.823.824. and 825. and precede other manifestations by years. They take the form of erythematous or violaceous nodules or plaques which may be widely distributed on the trunk and lower extremities. 821 Rarely paranasal or ulcerated palatal lesions are present. Any of the lesions may become ulcerated with eschar formation: this is more common with nodules on the leg.
The lung is the most commonly affected organ. The kidneys, liver, and central and peripheral nervous system may also be affected, although the skin is the most common extrapulmonary site.810.826. and 827.
Many patients when investigated are found to have defects in cytotoxic T-cell function and reduced numbers of CD8+ T cells. Consequently, LG occurs in patients with immunodeficiency states such as HIV/AIDS, Wiskott–Aldrich syndrome, and following organ transplantation. 819 There are overlap features with post-transplant lymphoproliferative disorders (PTLD) as both are B-cell lymphoproliferative disorders associated with EBV. The infiltrates of PTLD are ‘T-cell poor’ compared to the T-cell-rich infiltrates of LG. 828
Lymphomatoid granulomatosis has been reported in association with imatinib therapy, which can inhibit T-cell receptor mediated T-cell proliferation and activation, 829 and complicating other hematologic neoplasms associated with abnormalities of T-cell function. 830
Onset is usually in early middle age: it has rarely been reported in children. 831 In older studies there was a rapid downhill clinical course with a median survival of 14 months. 826 Death was usually from respiratory failure often associated with the development of an overt large B-cell lymphoma in the lungs. 817 Currently, longer survivals have been reported with multiagent chemotherapeutic and interferon therapy.811. and 814. Spontaneous regression of lesions may occur. 811
Histopathology
There is a polymorphous proliferation with perivascular, periappendageal, and perineural accentuation. 820 Sweat glands are particularly involved. 823
The infiltrate is composed of a mixture of small lymphocytes and a variable number of large lymphocytes with vesicular nuclei and prominent nucleoli which may resemble immunoblasts; some multinucleate forms resemble Reed–Sternberg cells.811. and 821. These cells are absent or rare in grade 1 lesions, are more prominent in grade 2 lesions, and form a major part of the infiltrate in grade 3 lesions which have features of diffuse large B-cell lymphoma. There are variable numbers of reactive histiocytes and eosinophils; neutrophils are present in ulcerated lesions. Occasionally, granulomas are present in skin lesions despite reports to the contrary in the lung literature (Fig. 41.17). As well as being angiocentric, the infiltrate is angioinvasive. Both arteries and veins may be affected. Cells invading vessel walls are predominantly small lymphocytes and histiocytes with fewer large atypical cells in low-grade lesions (Fig. 41.18). 811 Large atypical cells and fibrinoid necrosis of the vessel wall are seen in higher-grade lesions. Tissue necrosis is sometimes conspicuous in higher-grade lesions. Marked fibroblastic proliferation may also be present.
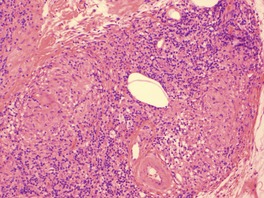 |
| Fig. 41.17 Lymphomatoid granulomatosis. There are granulomas in the infiltrate in this case. (H & E) |
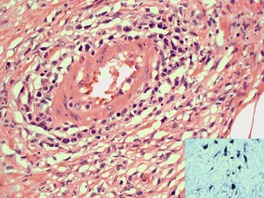 |
| Fig. 41.18 Lymphomatoid granulomatosis with many large, atypical lymphocytes in the wall of a vessel. (H & E) The inset shows positive staining of lymphocytes with EBV in-situ hybridization. |
Sometimes the histology of skin lesions may not be diagnostic, despite the presence of typical changes in other organs.
The differential diagnosis includes Wegener’s granulomatosis (WG), which only displays the classic combination of necrotizing granulomatous inflammation and a necrotizing vasculitis in a minority of cases (see p. 239). Atypical large lymphocytes are not seen in WG but they may also be absent in low-grade lesions of LG. Neutrophils are not usually seen in LG without necrosis and ulceration.
The presence of a largely histiocytic infiltrate with a perineural distribution may mimic leprosy on biopsies. 821
Immunohistochemistry and cytogenetics
The neoplastic cells are B cells and are CD20+, sometimes CD43+, and CD3−. They are variably positive for CD30. The atypical B cells are EBV positive. EBV intranuclear early RNA (EBER) can be demonstrated by in-situ hybridization techniques. 811 EBV+ cells, like atypical large B cells, are few or absent in grade 1 lesions, which may make the diagnosis difficult. EBV+ cells are said to be less frequently identified in skin lesions than lung lesions. Clonal rearrangement of immunoglobulin heavy-chain gene can be demonstrated in some cases. 811 The T cells are CD3+, and CD4+ cells predominate over CD8+cells.
PRECURSOR HEMATOLOGIC NEOPLASM
Only one entity will be considered under this heading.
Blastic plasmacytoid dendritic cell neoplasm
The current designation of this rare entity in the most recently published WHO/EORTC classification is CD4+/CD56+ hematodermic neoplasm (blastic NK-cell lymphoma) but it has been changed to blastic plasmacytoid dendritic cell neoplasm in the most recent WHO classification as the neoplastic cells share the markers for plasmacytoid dendritic cells.834.835.836.837.838. and 839. It was originally defined as an acute leukemia or cutaneous neoplasm that co-expressed CD4 and CD56 without specific markers for myeloid, B, T, or NK cells. As skin involvement usually precedes a leukemic phase it may represent a form of aleukemic leukemia cutis derived from precursor cells. 838
It most commonly affects adults (median age 67 years) with a male:female ratio of 2:1. 636 It has also been reported in children and adolescents.840.841.842. and 843.
It presents as skin lesions in 90% of cases but there is rapid spread systemically to many other sites including blood, bone marrow, lymph nodes, spleen, liver, and central nervous system.834.837.844.845.846. and 847. Skin lesions may be localized or disseminated and take the form of erythematous or bruise-like, red or purple patches, papules, nodules, and tumors.
This is an aggressive neoplasm with a poor prognosis. The median survival is 14 months. 636 Children and those who present with only skin lesions may have a slightly better prognosis.636. and 840.
Histopathology15.834. and 837.
There is an infiltrate of uniform medium-sized cells with little cytoplasm and nuclei with finely-dispersed cytoplasm and inconspicuous nucleoli in the dermis and/or subcutis (Fig. 41.19). The cells resemble myeloblasts or lymphoblasts. The epidermis is not involved and there is usually no necrosis, angioinvasion, or inflammatory cells in the infiltrate. ‘Rimming’ of adipocytes can be seen where the subcutis is infiltrated. 618
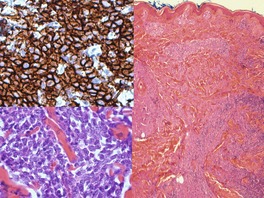 |
| Fig. 41.19 Blastic plasmacytoid dendritic cell neoplasm. There is a diffuse dermal infiltrate of blastic cells. (H & E) The cells are monotonous (lower inset); they are CD56+. (Top inset – immunoperoxidase stain) |
Immunohistochemistry and cytogenetics
This lesion was initially defined by the co-expression of CD4 and CD56 but this neoplasm can express markers of other lineages. The cells usually have a CD4+, CD56+, CD8–, CD2+/−, CD7+/–, CD45RA+ phenotype and are negative for CD3 and cytotoxic markers.834. and 848. The cells also express CD123 (the IL-3R alpha chain), a marker of plasmacytoid dendritic cells,849. and 850. TLC1 (lymphoid proto-oncogene), 851 and BDCA-2 (an immature dendritic cell marker). 852 CD68, TdT, and CD43836.851. and 853. are sometimes expressed. 636 CD56-negative cases have been reported. 848 Weak staining for CD33, a myeloid marker has also been described. 848 A rare, related condition, acute myeloid (or type 1) dendritic cell leukemia may present in the skin. In this condition myeloid markers are expressed as well as CD4, CD56, and CD123. 854
The TCR genes are usually germline but recently a rare variant with a T-cell gene rearrangement has been described. 855 Tumor cells are negative for EBV.
OTHER T/NK-CELL LYMPHOMAS THAT MAY INVOLVE THE SKIN
This group of lymphomas is not included with the other primary cutaneous lymphomas.
PRECURSOR T-LYMPHOBLASTIC LEUKEMIA/LYMPHOMA
This leukemia/lymphoma rarely presents in the skin and does so much less commonly than its B-cell counterpart.856. and 857. Both forms are more common in adolescent males. 858 No case of cutaneous involvement was recorded in a large series of cutaneous lymphomas in patients less than 20 years of age. 859 Cutaneous cellular infiltrates involving the dermis and subcutis consist of medium-sized cells with a high nuclear to cytoplasmic ratio, irregular nuclear contours and small nucleoli. The cells have a CD3+, CD5+, CD4+, CD8+, CD10+, and TdT+ immunoprofile. Cells are also positive for CD99. There is monoclonal TCR gene rearrangement.856. and 857.
T-CELL PROLYMPHOCYTIC LEUKEMIA
This is a rare aggressive leukemia in which there are cutaneous manifestations in many cases at the time of diagnosis, 24% in one series. 860 As well as a lymphocytosis there is lymphadenopathy and splenomegaly. There is a propensity for skin manifestations to involve the face, often symmetrically, but the limbs, and trunk may also be involved. There is often a diffuse infiltrated erythema. Edematous and purpuric lesions, plaques and nodules, and erythroderma may also be present.860. and 861.
Histopathology
There is a perivascular and periappendageal infiltrate involving the dermis, sometimes extending into the subcutis; the epidermis is spared. 860 The infiltrating cells are small to intermediate-sized lymphocytes with oval, reniform, and irregular nuclei having finely dispersed chromatin and a small single central nucleolus. There is a high nuclear/cytoplasmic ratio. The nucleus may be eccentrically placed. 861
Immunohistochemistry and cytogenetics
In T-cell prolymphocytic leukemia the cells are CD2+, CD3+, CD5+, and CD7+. Approximately two-thirds of cases are CD4+ but they may be CD4+/CD8+ or CD4−/CD8+.1. and 862. Cells also stain in a nuclear and cytoplasmic pattern for TCL-1 (T-cell leukemia oncoprotein). 862
ANGIOIMMUNOBLASTIC T-CELL LYMPHOMA (AITL)
This lymphoma was originally known as angioimmunoblastic lymphadenopathy with dysproteinemia (AILD) and was considered an abnormal immune reaction with B-cell hyperplasia. 863 It is now regarded as a T-cell lymphoma as most cases have clonal TCR gene rearrangements. 864 The pathogenesis of AITL is uncertain. It has been proposed that the disease begins as a primary monoclonal proliferation of T cells or is initially a polyclonal response to a drug or virus which then evolves into a monoclonal proliferation.865. and 866. It is characterized by generalized lymphadenopathy, hepatosplenomegaly, constitutional symptoms such as fevers, night sweats and weight loss, polyclonal hypergammaglobulinemia, and, in about 50% of cases, a skin rash.867. and 868. This often consists of a non-specific, generalized maculopapular rash resembling a viral exanthem or drug eruption. Lesions may precede or occur concurrently with systemic disease and are commonly on the trunk and limbs. Other cutaneous manifestations include erythematous plaques, nodules, and urticarial and purpuric lesions. Lesions may regress spontaneously or after corticosteroid therapy. It occurs most commonly in the elderly. The median survival in this condition is 11–30 months. 863Infections are common complications. A second nodal EBV-associated B-cell lymphoma may develop.1. and 869. Cutaneous B-cell lymphoma has also been reported in association with this condition. 870
Histopathology865
The histological appearance of lesions is variable: the most common pattern is a non-specific perivascular infiltrate of non-atypical lymphocytes with some vascular proliferation. 865 In some cases there is a sparse perivascular infiltrate of pleomorphic lymphocytes.
In those cases most recognizable as lymphoma, there is a variably dense perivascular and periappendageal infiltrate that may extend into the subcutis. Cellular infiltrates are polymorphous and include variable numbers of large atypical lymphocytes, medium and small lymphocytes and admixed histiocytes, plasma cells and eosinophils. Rarely, Reed–Sternberg-like cells are seen. 869 Proliferation of post-capillary venules is also seen in many biopsies, similar to the vascular proliferation seen in lymph nodes. Occasionally there is a leukocytoclastic vasculitis with extravasated red blood cells. An infiltrate with granulomas871 and one with necrotizing granulomas have been reported. 872
Immunohistochemistry and cytogenetics
The atypical neoplastic lymphocytes express T-cell antigens CD2, CD3, CD4, and CD45RO. Recently neoplastic T cells have been reported to show aberrant expression of CD10 in nodes and at extranodal sites.873. and 874. These cells also frequently express CXCL13, a chemokine normally expressed by follicular helper-T cells; it was demonstrated in 80% of skin biopsies in one series. 875
Overexpression of c-Maf (a member of the basic leucine zipper transcription factors belonging to the AP1 superfamily) has been demonstrated in the neoplastic T cells in frozen sections. 876
There are also CD20+ B cells in the background population of cells. EBV is detected in lymph nodes in the majority of cases of AITL but only in a few isolated lymphocytes. It has been reported only rarely in skin lesions.865.869. and 877. Clonal TCR gene rearrangements have been reported in 75% of lymph nodes and have also been reported in the skin, even in non-specific infiltrates. DNA microassay analysis has been performed on a limited number of cases. 878
PRIMARY SYSTEMIC ANAPLASTIC LARGE CELL LYMPHOMA
The primary systemic form of anaplastic large cell lymphoma (ALCL) can be further subdivided by whether the tumor cells express the anaplastic lymphoma kinase fusion protein (ALK) or not. ALK+ and ALK− cases have different clinical presentations and response to therapy. 879
ALK+ systemic ALCL occurs most commonly in the first three decades of life with a definite male predominance (M:F – 3:1). Extranodal involvement occurs in 60% of cases with the skin being the most common extranodal site (21% of cases). ALK− systemic ALCL is more common in older age groups and there is an almost equal sex incidence (M:F – 0.9:1). ALK+ cases have a better response to therapy and prognosis than ALK− cases. 37
Skin presentation may precede the detection of a systemic lymphoma. 880 An erythrodermic form has rarely been reported. 880 Primary systemic ALCL has been associated with acquired ichthyosis. 881
Histopathology
The common type of cells in ALCL are large lymphoid cells with chromatin-poor, horseshoe-shaped, or embryo-shaped nuclei, multiple prominent eosinophilic nucleoli and abundant cytoplasm, often with a pale paranuclear hof. These cells are sometimes referred to as ‘hallmark’ cells and are seen in all cytological forms of ALCL. In most cases the cells vary in size and shape but they are sometimes relatively monomorphous. 882 The common type of ALCL comprises 60–70% of all cases. Multinucleate cells resembling Reed–Sternberg cells may also be seen.220. and 883. The small cell variant has a mixture of small, medium, and large lymphoid cells. Large cells often surround vessels. Small cells are often CD30−. 884 In the lymphohistiocytic variant there is a heavy histiocytic infiltrate which may mask the smaller neoplastic cell population of lymphoid cells. Cells are often smaller than those in common ALCL. 885 The small cell type and the lymphohistiocytic type represent approximately 5% and 15% of cases, respectively, and are predominantly in the pediatric group.886. and 887. The giant cell variant is characterized by many multinucleate cells. 220 A sarcomatoid variant resembles a soft tissue sarcoma and contains large atypical, often spindle-shaped cells. 888 Eosinophil-rich and neutrophil-rich variants have also been described.889.890.891. and 892. A rare signet-cell type has also been reported. 893
In cutaneous involvement, there may be a diffuse dermal infiltrate or an infiltrate restricted to perivascular and periappendageal sites. 894 The epidermis is spared.
Immunohistochemistry and cytogenetics
Tumor cells by definition are predominantly CD30+ with staining of the cell membrane and Golgi zone. ALK-1 is positive in 60% of cases. The majority are EMA+. CD45 is expressed in 60% of cases. T-cell antigens CD3, CD2, and CD45RO are seen in approximately 50% of cases. A minority are CD4+ or CD8+. CD56, TIA-1, granzyme B, and perforin may be expressed.220.883. and 895.
ALK protein is absent in normal tissues except the brain, and expression in other tissues indicates anomalous ALK expression usually in the form of nucleophosmin (NPM–ALK) fusion protein associated with a t(2;5)(p23;q35) translocation. Other translocations such as t(1;2)(q21;p23), t(2;3)(p23;q21) and inversion2 (p23;q35) have also been reported. Immunohistochemistry, FISH, and ALK gene rearrangement studies are equally effective in identifying ALK+ cases.
As stated above, the skin lesions in primary cutaneous ALCL are almost invariably ALK negative and do not have the t(2;5) translocation, although rare ALK+ cases with or without the t(2;5) translocation have been reported.505.896.897.898. and 899. CD44 (a cell surface adhesion molecule) is highly expressed in both systemic and cutaneous forms of ALCL whereas the variant form CD44v6 is expressed in 90% of the systemic form but in only 50% of the cutaneous form. 529
Approximately 90% of systemic ALCL exhibit clonal TCR gene rearrangements whether or not they express T-cell antigens. The remainder show no rearrangements of TCR or Ig genes. 900
EBV is not expressed in cells. 901
INTRAVASCULAR T- AND NK-CELL LYMPHOMA
Cases of intravascular lymphoma of T-cell or NK-cell type are very rare.638.794.902.903. and 904. In one series of intravascular lymphoma they represented 2.6% of cases. 761 Clinical presentation is similar to those with the B-cell form, with the skin being the predominant presenting site, followed by the central nervous system. The trunk and extremities are predominantly involved. 794 Unlike the B-cell form there is a common association with EBV. The prognosis is very poor and worse than that of the conventional lymphoma of that type. 902
Histopathology
Vessels in the dermis and subcutis contain intermediate to large atypical lymphocytes. Sometimes the vessels are surrounded by smaller, non-neoplastic lymphoid cells.794. and 903. Thrombi may be present in vessels. 638 Apoptotic bodies may also be identified in vessel lumina.
Immunohistochemistry and cytogenetics
The phenotype is variable and can be of a NK- or T-cell type. NK cell tumors may have a phenotype similar to the NK/T-cell lymphoma, nasal type. T-cell tumors may express cytotoxic markers. 904 T-cell tumors have clonal rearrangement of TCR genes; in NK lymphoma TCR are germline. Cases may have an anaplastic large cell phenotype (CD30+). EBV is present in more than half the cases reported. 794
AGGRESSIVE NK-CELL LEUKEMIA
Aggressive NK-cell leukemia, also known as aggressive NK-cell leukemia/lymphoma905 is a very rare disorder in Western countries, most reports coming from Japan.632.906. and 907. It occurs in a younger age group than NK/T-cell lymphoma, nasal type (median age of 30) in reported cases, 906 but there is overlap between these two conditions. It is characterized by an aggressive disease with fevers and hepatosplenomegaly as well as lymph node and bone marrow involvement. There are circulating atypical large lymphoid cells with azurophilic granules. Skin involvement is said to be rare908 but in one review of the literature it was present in almost 50% of cases. 906
Histopathology
Features overlap with those of extranodal NK/T-cell lymphoma, nasal type. There is a dermal infiltrate of mononuclear cells which are arranged in an angiocentric pattern with angioinvasion and destruction. 909
OTHER T/NK-CELL LYMPHOMAS AND LEUKEMIAS
T- and NK-large granular cell leukemias very rarely present as specific infiltrates in the skin. 910 Other non-specific skin manifestations are much more common.911. and 912. There are circulating lymphocytes with abundant cytoplasm and coarse azurophilic granules. The majority are of T-cell type (80%). 910 The common immunophenotype of the T-cell form is CD3+, CD4−, CD8+ with variable CD56 and TIA-1. There are clonal TCR gene rearrangements. The NK variant is CD3− with no TCR gene rearrangements. There is an indolent course in most cases.
Cutaneous lesions of Lennert’s lymphoma (lymphoepithelioid T-cell lymphoma) have rarely been reported. 913
An indolent CD8-positive T-cell lymphoma occurring on the ear has been reported. 914 In these cases there is a dermal infiltrate of monomorphous medium-sized cells with an irregular nucleus, small nucleoli, and minimal cytoplasm. There is no epidermotropism. The cells have a CD3+, CD8+, TIA-1, CD45RA, βF1 immunoprofile.
There remains a group of peripheral T-cell lymphomas, unspecified which present in the skin. They have been divided into small/medium sized T-cell lymphomas and CD30− large T-cell lymphomas. Apart from a small group of CD3+, CD4+, CD8− small/medium-sized T-cell lymphomas with solitary or localized skin lesions (see above), this group of lymphomas has an unfavorable prognosis with an overall 5-year survival of 20%. 663
OTHER B-CELL LYMPHOMAS THAT MAY INVOLVE THE SKIN
PRECURSOR B-LYMPHOBLASTIC LEUKEMIA/LYMPHOMA
A small number of patients present primarily as a lymphoma (PBLL) rather than a leukemia (PBLA). Unlike precursor T-lymphoblastic lymphoma (PTLL), which commonly involves lymph nodes and mediastinum, PBLL usually involves extranodal sites, the skin being the most common site of extranodal presentation. Cutaneous presentation of PBLL occurs in children, including the very young, 909 and adults,857.915. and 916. with the head and neck region being the most commonly involved sites. The lesions are erythematous or violaceous papules or nodules. PBLA may present with urticarial skin lesions. 917 Although systemic spread is demonstrated when staging is undertaken, there are reported cases limited to the skin. 918 Aggressive chemotherapy may prevent progression to leukemia, 918 and PBLL appears to have a better prognosis than PBLA.
Histopathology856. and 918.
The neoplastic infiltrate involves the dermis and subcutis with sparing of the epidermis. There is commonly a ‘starry-sky’ pattern with gaps in the infiltrate containing macrophages with cell debris. A vague nodular pattern can be produced by collagen compartmentalization. Crush artifact and the ‘Azzopardi effect’ (basophilic smearing of collagen fibers) are common in biopsies. The cellular infiltrate is monomorphic and consists of medium-sized lymphoid cells with round or convoluted nuclei, inconspicuous nucleoli, and little cytoplasm. Numerous mitotic figures can be present, including atypical forms.
Immunohistochemistry and cytogenetics
The neoplastic cells express the B-cell marker CD79a and less commonly CD20. They are CD10+, TdT+ (terminal deoxynucleotidyl transferase), CD99+, and bcl-2+ in most cases, but not always. 909 CD99 is particularly useful in distinguishing this lymphoma from other B-cell lymphomas. Cells are Pax5+ whereas the cells in T-lymphoblastic leukemia/lymphoma are negative. 759 The cytogenetics of this condition are complex and are prognostically important. Rearrangement of the MLL gene and hyperdiploidy have been reported in cutaneous lesions. 916
CHRONIC LYMPHOCYTIC LEUKEMIA/SMALL LYMPHOCYTIC LYMPHOMA (B-CLL)
The specific infiltrates of B-CLL include localized or generalized, erythematous papules, plaques, nodules, and large tumors. Unusual presentations include involvement of parts of the face and ears. 919 Reflecting the fact that B-CLL occurs predominantly in older adults, 1 the mean age of presentation in one series was 66 years. 920 Uncommonly, skin lesions may represent the first sign of the disease.920.921. and 922. Lesions may present in the scars of herpes zoster or varicella, or herpes simplex lesions.923.924. and 925. It may also occur at sites typical for lymphoid hyperplasia associated with Borrelia burgdorferi infection, such as the nipple and scrotum. 926 Occasionally B-CLL and mycosis fungoides occur together. 927 Acute myelogenous leukemia cutis has also been described in association with B-CLL. 928
The 5-year survival of B-CLL with cutaneous lesions is 66%. Transformation to large B-cell lymphoma (Richter’s syndrome) is associated with a poor prognosis.
Histopathology
Patterns of dermal infiltration include perivascular and periappendageal, nodular, diffuse, and band-like. 920 The cellular infiltrate consists of a monomorphous population of small lymphocytes. Proliferation centers, which are pale areas containing larger prolymphocytes and paraimmunoblasts are uncommon. 920 Varying numbers of larger cells resembling centroblasts and immunoblasts are present in cases undergoing transformation to a large B-cell lymphoma. There may be epidermal changes of acanthosis and ulceration. Other reactive cells including eosinophils, neutrophils, histiocytes, and plasma cells may be present. 929
The leukemic infiltrate may surround epithelial neoplasms such as basal cell carcinoma and squamous cell carcinoma (Fig. 41.20).930. and 931. The pathologist should be alerted to this possibility when there is a dense basophilic dermal infiltrate evident with the naked eye in a slide of an excised skin lesion, particularly if it is present in a number of lesions that have been excised.
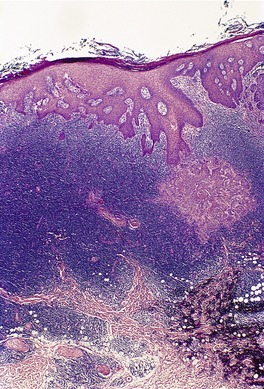 |
| Fig. 41.20 Basal cell carcinoma associated with an infiltrate of B-cell chronic lymphocytic leukemia. (H & E) |
MANTLE CELL LYMPHOMA
The majority of cases classified as mantle cell lymphoma (MCL) in the skin are secondary to nodal disease. 932 Secondary skin involvement has been reported in up to 17% of cases of advanced disease. 933 Cutaneous lesions may be the first manifestation of the disease. 611
Histopathology
As defined in the WHO classification, this is a lymphoma composed of small to medium-sized lymphocytes with irregular or cleaved nuclei with dispersed chromatin, inconspicuous nucleoli, and scant cytoplasm most closely resembling centrocytes/cleaved follicular center cells. 934 A ‘blastoid’ variant with cells resembling lymphoblasts with larger nuclei and dispersed chromatin has also been described with skin involvement. 935
In two possible cases of primary MCL, there were nodular infiltrates of the characteristic cells surrounding atrophic germinal centers in a mantle zone pattern; the dermis and subcutis were involved with sparing of the epidermis. 936 Plasma cells were present at the periphery of the nodules.
Secondary involvement most often takes the form of a perivascular or periappendageal infiltrate or a diffuse infiltrate that spares the epidermis but may extend into subcutis. The infiltrate is reported to have the blastoid subtype frequently. 937
Immunohistochemistry
The neoplastic cells are CD20+, CD5+, CD43+, CD10−, CD23−, and bcl-6−. Some cases are CD5−. Rare cases have aberrant bcl-6 expression. Most have nuclear staining for cyclin D1.938 The expression of this nuclear protein is due to overexpression of the PRAD1 gene as a result of the translocation t(11;14)(q13;q32) involving the immunoglobulin heavy-chain locus and the bcl-1 locus. The translocation is characteristic of MCL938. and 939. and can be demonstrated by FISH, PCR, or conventional cytogenetics.937. and 940.
PRIMARY EFFUSION LYMPHOMA
Primary effusion lymphoma is a rare lymphoma of large B cells that presents as serous effusions without an associated tumor mass. In most cases, but not all, it is associated with HHV-8 infection. It is most commonly seen in young patients with HIV/AIDS941 but can also be seen in transplant recipients and elderly men in areas with a high prevalence of HHV-8. It has rarely been reported secondarily in the skin. 942 In a reported case there was a diffuse infiltrate of large B cells (CD20+, CD79a+, CD3−) in the dermis and subcutis. 942
LYMPHOPLASMACYTIC LYMPHOMA/WALDENSTRÖM’S MACROGLOBULINEMIA (LL/M)
The current WHO classification defines this rare entity as a lymphoma of small B lymphocytes, plasma cells, and plasmacytoid cells associated with an IgM paraprotein. 943 The immunoprofile of the component cells is usually CD5−, CD10−, and CD23−.943. and 944. There appears to be some overlap with marginal zone B-cell lymphoma. 944 The term ‘immunocytoma’ is confusing and has been used for LL/M as well as marginal zone lymphoma in the skin. Cases with cutaneous presentation have been described. 945
γ Heavy-chain disease is classified as a variant of LL/M in the current WHO classification. 946 It is a biochemical expression of a mutant clone of B cells which produces abnormal, incomplete γ heavy chains, devoid of light chains. 947 It is usually associated with a lymphoplasmacytic proliferative disorder, although the associated clinical and histological findings are varied. 948 Cutaneous involvement is rare, being present in less than 5% of the reported cases. 947 The usual skin lesions are erythematous infiltrated plaques and nodules on the trunk and extremities. Livedo reticularis and digital necrosis caused by associated necrotizing arteritis have been reported. 949
Histopathology
In the few reported cases of γ heavy-chain disease with cutaneous involvement there has been a dermal infiltrate of lymphoplasmacytic cells, immunoblasts, and mature plasma cells. 947 In one case, vascular proliferation and the presence of eosinophils led to an initial diagnosis of angiolymphoid hyperplasia with eosinophilia.948. and 950. Sometimes the cells have a conspicuous periadnexal distribution.
BURKITT AND BURKITT-LIKE LYMPHOMA
This lymphoma has rarely been reported in the skin as a secondary lesion. 951 It may present in the skin around nodal biopsy sites, 952 and has been reported in a background of HIV/AIDS.663. and 953. In Burkitt lymphoma (BL) there is an infiltrate of monomorphous medium-sized cells with round nuclei and multiple central nucleoli. There are numerous mitotic figures, and macrophages with phagocytosed apoptotic bodies are frequent, producing the characteristic ‘starry-sky’ appearance. In Burkitt-like lymphoma (BL-L) the cells are slightly larger and more pleomorphic; there are overlapping features with diffuse large B-cell lymphoma. Both BL and BL-L have a proliferation fraction approaching 100% (with Ki-67). The cells are CD20+, CD10+, CD43+, and bcl-6+. BL is characterized by a translocation of the MYC proto-oncogene on chromosome 8 (t(8;14) or t(2;8)).
PLASMACYTOMA AND SECONDARY MYELOMA
Although plasmacytomas are now regarded as variants of lymphoma, they have been considered in Chapter 40, p. 943, with other diseases of plasma cells.
OTHER LYMPHOMAS
HODGKIN LYMPHOMA
Infiltration of the skin is usually a late manifestation of Hodgkin lymphoma (HL), and develops in from 0.5 to 3.4% of patients according to old literature.954.955. and 956. Current experience suggests it has decreased in incidence because of improved therapy for nodal disease. 957 Lesions often occur in a localized area of the trunk or on the proximal part of the extremities in the vicinity of involved lymph nodes, suggesting retrograde lymphatic spread.954. and 958. Hematogenous and in-continuity direct spread are other potential routes of dissemination. Direct skin invasion from nodes may mimic scrofuloderma. 959
Cutaneous lesions may be papules, nodules, or plaques, although rare clinical presentations have included multiple ulcers on the scalp, 960 a bullous eruption, 961 and widespread lesions. Paradoxically, two of the cases with widespread deposits had a more prolonged survival than is usually seen with cutaneous disease. 962 Spontaneous regression of the skin lesions has been reported. 963
Skin lesions may rarely be the initial manifestation of HL with lymph node involvement appearing some months or years later.964.965.966.967.968. and 969. Primary cutaneous HL is a controversial entity, although rare cases appear to exist.970. and 971. Some cases previously reported may represent lymphomatoid papulosis or anaplastic large cell lymphoma. Cases of lymphomatoid papulosis followed by HL have been reported.574. and 575. Primary cutaneous HL may pursue a benign course, although up to 50% of patients develop nodal or systemic disease.970. and 971.
A variety of non-specific skin manifestations are commonly found in the skin in HL. These include pruritus, herpes zoster, and acquired ichthyosis. Less common associations have included urticaria, eczema, erythema multiforme, erythema nodosum, drug eruptions, bullous pemphigoid, dermatitis herpetiformis, pemphigus, acquired epidermolysis bullosa, follicular mucinosis, alopecia, lymphedema, dermatomyositis, and granuloma annulare. 972 Numerous examples of concurrent HL and mycosis fungoides have been reported. Rarely, mycosis fungoides and cutaneous HL have been reported together.968. and 973.
The WHO classification divides HL into classical HL (including nodular sclerosis, lymphocyte-rich classical, mixed cellularity, and lymphocyte depletion subtypes) and nodular lymphocyte predominance HL, which is now regarded as a separate clinicopathological entity. 2
The neoplastic cells of HL have been shown to be clonally derived from germinal center B cells that have lost the capacity to produce or express immunoglobulin.974. and 975. In addition, many epidemiological and molecular studies suggest that Epstein–Barr virus (EBV) plays a significant etiological role in HL.976.977. and 978. In-situ hybridization studies for EBV have demonstrated abundant viral transcripts in Reed–Sternberg cells. 971
Histopathology
There is a diffuse infiltrate of cells involving the dermis and subcutis. A grenz zone is usually present. Skin appendages and blood vessels are frequently invaded. 965 There is a mixed cellular infiltrate which includes mononuclear variants of Reed–Sternberg cells and variable numbers of classic Reed–Sternberg cells in a background of small lymphocytes, eosinophils, and sometimes neutrophils (Fig. 41.21). Fibrosis is another common feature. The histology may resemble that of nodular sclerosis or mixed cellularity types; lymphocyte depletion may be present in advanced stages. 954 Non-specific changes reported in the skin in association with the lymphomatous infiltrate include epithelioid granulomas, 979 and palisading necrobiotic granulomas. 980
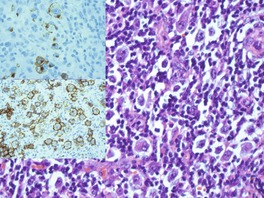 |
| Fig. 41.21 Cutaneous Hodgkin lymphoma. Numerous Reed–Sternberg cells are present. (H & E) The cells are CD15+ (top inset), and CD30+ (lower inset) – immunoperoxidase stains for CD15 and CD30. |
The histology of HL, lymphomatoid papulosis, and anaplastic large cell lymphoma has many features in common. Neutrophils and eosinophils have been reported in lesions from all these conditions and are not a discriminating feature. Reed–Sternberg-like cells may be seen in lymphomatoid papulosis and anaplastic large cell lymphoma, on occasions.
Immunohistochemistry and cytogenetics
Reed–Sternberg cells and mononuclear variants are CD30+, CD15+ in most cases, and are CD45−, CD45RO−, CD43−, and EMA−. Occasional cases are CD15−. 955 Cells may occasionally express B-cell markers CD20, Oct-2, and Bob.1 but in a patchy fashion. 981 They are usually Pax5+, whereas the cells of anaplastic large cell lymphoma do not stain. 759 EBV may be demonstrated in cells by immunohistochemistry for LMP-1 or by in-situ hybridization for EBER. 982 The background population of small lymphocytes are predominantly CD3+, CD4+ T cells.
Large cells in anaplastic large cell lymphoma and lymphomatoid papulosis are usually CD30+, CD15−, and CD45 may be negative in some cases. 37 They may be EMA+ more commonly in the primary systemic form than in the cutaneous form and lymphomatoid papulosis. ALK-1 may be positive in the systemic form, rarely in the cutaneous form and never in HL. 220
No cytogenetic studies have been performed on cutaneous disease. Studies on HL in lymph nodes have shown clonal rearrangements of immunoglobulin genes in tissue occasionally, but in 98% of isolated Reed–Sternberg cells. 981
CUTANEOUS INFILTRATES OF LEUKEMIAS
Some of the leukemias have already been discussed with their lymphomatous counterparts. A discussion of myeloid leukemia and its subtypes follows.
MYELOID LEUKEMIAS, MYELOPROLIFERATIVE DISEASES, AND MYELODYSPLASTIC SYNDROMES
Cutaneous infiltrates have been described in many forms of acute myeloid leukemia, chronic myeloproliferative diseases (such as chronic myeloid (myelogenous) leukemia), and in the setting of myelodysplastic syndromes and myeloproliferative syndromes as a sign of transformation to an acute leukemia.983.984.985.986.987.988.989.990.991.992.993. and 994.
Leukemia cutis, myeloid sarcoma, granulocytic sarcoma, and chloroma are all terms that have been used for leukemic infiltrates in extramedullary sites including the skin. 995‘Chloroma’ refers to tumor masses that are green in color on exposure to light due to the presence of myeloperoxidase. 996
Myeloid leukemic infiltrates can take many forms clinically but mostly present in adults as single997 or multiple macules, papules, nodules, or plaques, often with a red/brown or violaceous color in no particular distribution. 989 Unusual presentations include a stasis dermatitis-like eruption,998. and 999. a chilblain-like eruption,1000. and 1001. cutaneous hyperpigmentation, 1002 and macrocheilia. 1003 Leukemic infiltrates can occur in children and are sometimes congenital,1004. and 1005. or precede demonstrable blood leukemia (aleukemic). 1006 Spontaneous regression in a congenital case has been reported. 1007
Oral lesions, gum hypertrophy, and macroglossia have also been reported and are common in some subtypes, particularly acute monocytic and myelomonocytic forms.989. and 1008. Lesions may precede the development of frank leukemia, often by months,1009.1010.1011.1012. and 1013. and rare cases with long survivals (usually after chemotherapy) without progression to leukemia have been reported.1014.1015. and 1016. Generally the prognosis is poor, however.985. and 1017. It may be the first sign of relapse after bone marrow transplantation for acute myeloid leukemia. 1018
Commonly used classifications of this group of conditions include the French-American-British classification (FAB classification) 1019 and the WHO classification. 1020 The incidence of cutaneous involvement in acute myeloid leukemia is in the range of 2–20%, 991 and in chronic myeloid leukemia, 0–4%. 1021 Of acute myeloid leukemia, cutaneous involvement is most common in FAB type M4 (acute myelomonocytic leukemia) and FAB type M5 (acute monocytic leukemia) but is seen in most subtypes.989.991. and 1022. Skin infiltration is uncommon in chronic myelomonocytic leukemia.1023. and 1024. Rare leukemias such as myeloid/NK cell precursor leukemia may present in the skin. 1025
Histopathology989
Leukemic infiltrates may take the form of perivascular and periappendageal infiltrates or confluent sheets of cells that involve the dermis and often the subcutis. There may be destruction of appendages. Sometimes there is percolation between collagen bundles in an ‘Indian file’ pattern reminiscent of the pattern seen in lobular carcinoma of the breast (Fig. 41.22). There may also be concentric layering of cells around vessels and adnexal structures.
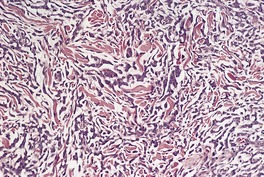 |
| Fig. 41.22 Cutaneous infiltration by myeloid blasts in a patient with acute myeloid leukemia. (H & E) |
The cell types present depend on the subtype of myeloid leukemia. In FAB M1 and M2 types (acute myeloblastic leukemia), the cells are predominantly atypical myeloblasts and myelocytes (medium to large mononuclear cells with a slightly eccentric basophilic nucleus, a single nucleolus, and a small amount of cytoplasm). FAB M4 and M5 types (acute myelomonocytic and monocytic leukemias) have infiltrates composed of medium-sized atypical round to oval monocytoid cells with indented or kidney-shaped nuclei.
In chronic myeloid leukemia the infiltrate is more pleomorphic with immature and mature granulocytes (myelocytes, metamyelocytes, eosinophilic metamyelocytes, and segmented neutrophils). 989 Metamyelocytes with large eosinophilic granules (eosinophilic metamyelocytes) are a useful clue that an infiltrate is of myeloid lineage and not lymphoma (Fig. 41.23).
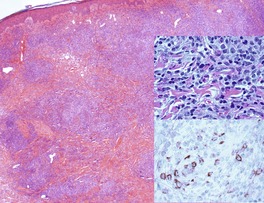 |
| Fig. 41.23 Acute myeloid leukemia. There is a heavy infiltrate of blasts, some of which contain conspicuous eosinophilic granules (top inset). Some cells are positive for myeloperoxidase (bottom inset). (H & E and immunoperoxidase stain) |
Mitotic figures and apoptotic bodies are often seen. Other uncommon histological features include the presence of Langhans type giant cells1026 and leukemic vasculitis with leukemic cells in the walls of vessels sometimes with fibrinoid necrosis.1027. and 1028.
A positive naphthol AS-D chloroacetate esterase (NSAD) reaction was present in 64% of infiltrates in acute myeloid leukemia and 72% in chronic myeloid leukemia in one series. 989
Leukemic infiltrates have been reported in basal cell carcinoma, 1029 psoriasis, 1030 and Sweet’s syndrome. 1021
Immunohistochemistry and cytogenetics
The immunoprofile is determined by the type of myeloid infiltrate. Overall CD68 and lysozyme are the most sensitive immunostains for the detection of myeloid leukemic infiltrates, but they are not lineage specific. 991
Myeloperoxidase, CD45, CD74, HLA-DR, CD43, and Mac387 are expressed in a large percentage of cases. Myeloperoxidase is less useful in cases with monocytic differentiation. Other markers that may be present in a smaller percentage of cases include CD56, CD34, CD117, and CD15.989.990.991. and 1012. CD30 is expressed rarely. 1031
LYMPHOID HYPERPLASIAS MIMICKING PRIMARY LYMPHOMA
This category includes a variety of benign lymphoid proliferations which simulate cutaneous lymphoma clinically, but particularly histopathologically. The term ‘pseudolymphoma’ has been used for some of these conditions but is best avoided since it suggests a diagnostic category and several conditions included under this umbrella do not pose difficulties in their distinction from true lymphoma. Lymphomatoid papulosis was regarded in the past as a pseudomalignancy but is now classified with other cutaneous lymphomas (see above). 15
Traditionally, this group of conditions has been divided into B-cell and T-cell types based on the pattern and composition of the cellular infiltrate. 1032 The histological pattern may take the form of a superficial band-like infiltrate in the upper dermis (the so-called ‘T-cell pattern’) or a nodular or diffuse infiltrate in the dermis and sometimes subcutis (the so-called ‘B-cell pattern’). This is an artificial distinction since predominantly T-cell proliferations may have a nodular pattern of dermal infiltration. In cutaneous lymphoid hyperplasia of the B-cell pattern, T cells are in fact more common than B cells. 1033
Interpretation of apparent clonal rearrangement of TCR genes or IgH genes by PCR must be interpreted with caution and correlated with the histological appearance. The problem of so-called ‘pseudoclones’ (a clone that on double or triple testing appears in a different position) has been addressed. The importance of duplicate or triplicate tests for clonality by PCR has been stressed to exclude overdiagnosis of clonality. 1034
CUTANEOUS LYMPHOID HYPERPLASIA SIMULATING B-CELL LYMPHOMA
A plethora of terms has been used for this form of lymphoid hyperplasia: B-cell pseudolymphoma, B-cell cutaneous lymphoid hyperplasia, lymphadenosis benigna cutis, lymphocytoma cutis, and cutaneous lymphoplasia.1035. and 1036. These lesions usually present as asymptomatic red-brown or violaceous papules or nodules varying in diameter from 3 mm to 5 cm or more. 1037 They may be solitary, grouped or numerous and widespread. 1038
The most common sites of involvement include the face (cheeks, nose, and earlobe), chest, and upper extremities. 584 A xanthelasma-like presentation has been reported. 1039 Females are affected more often than males. Lesions may resolve spontaneously after months or years, but there is a tendency for some to recur.1037.1038. and 1040. None of the clinical features allows reliable distinction from cutaneous lymphoma.
In most cases the cause is unknown. Various stimuli are reported to induce this form of lymphoid hyperplasia, 1041 including tick and other arthropod bites and stings,1042. and 1043. Hirudo medicinalis (leeches), 1044 gold earrings and gold injections,1045. and 1046. cobalt, 1047 zinc, tattoos (particularly the red areas),1048.1049. and 1050. a chronic draining sinus, 1051 ingestion of drugs such as phenytoin sodium, 1052 methotrexate, 1053 doxepin, and clozapine, 1054 hair coloring products (paraphenylenediamine), 1055 immunization,1056. and 1057. and specific immune therapy. 1058 Multiple cutaneous lesions have developed following allergen injections given for hyposensitization.1059. and 1060.
In Europe, cutaneous lymphoid hyperplasia (lymphocytoma cutis) has been associated with Borrelia infection and is said to be the most common cause in endemic areas.1061.1062. and 1063. This is a rare event in North American Lyme disease. 1064 In this setting, lesions occur particularly on the earlobes, areolae of the nipples, 1065 and scrotum, 89% of cases in one series. 1063 It has been described in association with acrodermatitis chronica atrophicans. The prevalence of Borrelia-associated cutaneous lymphoid hyperplasia has been reported to be from 0.6% to 1.3% of cases where there has been a clinical and/or serological diagnosis. 1066 It is more common in women than men and is seen in a wide age range which includes children.1063. and 1064.
The use of immunohistochemistry and molecular techniques, and a better understanding of the histopathological subtypes of cutaneous lymphoma has made the distinction of cutaneous hyperplasia from low-grade cutaneous B-cell lymphomas more reproducible. The significance of a clonal proliferation of B cells in a histologically equivocal lesion remains controversial. The presence of a clonal B-cell population did not correlate with the subsequent behavior of lesions in one series. Occasionally both B- and T-cell clonality has been demonstrated in the same case.1065.1067. and 1068. A monoclonal population of B cells was identified in 14% of cases diagnosed as pseudolymphoma on histological and clinical grounds in one study. 1069 Another study proposed that there is a spectrum of cutaneous lymphoid proliferations that includes some lesions in which occult dominant clones can be identified by PCR or Southern blot analysis, PCR being a more sensitive method of detection. In this study dominant clones of B or T cells were demonstrated in 61% of cases. In time, 4% of cases progressed to overt cutaneous B-cell lymphoma. 1054 It is still not clear in what proportion of cases transformation to cutaneous lymphoma occurs and if ‘transformation’ may in fact represent undiagnosed early cutaneous lymphoma. 1070 Transformation to a malignant lymphoma has been reported to occur in 25% of presumed benign lymphoid hyperplasias where a clonal B-cell population has been identified, compared with 5% of cases without demonstrable clonality. 1069
Histopathology
The histological appearances are varied and there is considerable overlap with primary cutaneous follicle center cell lymphoma and primary cutaneous marginal zone B-cell lymphoma. There is a variably dense infiltrate which may have a perivascular and periappendageal distribution or be more diffuse. The epidermis is usually spared but some small lymphocytes may be seen in the epidermis. 1063 A ‘top heavy’ cellular infiltrate is more common than a ‘bottom heavy’ one but is not specific (Fig. 41.24). The infiltrate may extend into the subcutis. In reactions at the site of vaccination, the subcutis is predominantly affected with little dermal involvement.1056. and 1057. Lymphoid follicles are present in many but not all cases, and well-developed mantle zones are seen in a minority which may present a difficulty in distinguishing hyperplasia from follicle center cell lymphoma. The composition of follicles is different from that seen in follicle center cell lymphoma in that centroblasts are more prominent and tingible body macrophages are always present. 1063 Stains for follicular dendritic cells may demonstrate follicular aggregates without germinal centers. Fusion of irregular follicle centers may be seen to produce a pattern similar to that in diffuse large B-cell lymphoma. 1071 Well-formed lymphoid follicles with germinal centers are seen in lesions following vaccination.1056. and 1057.
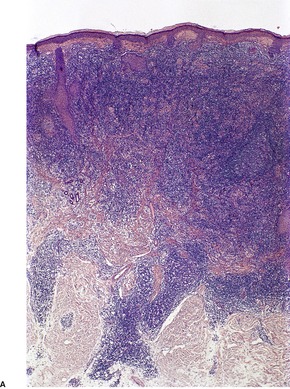 |
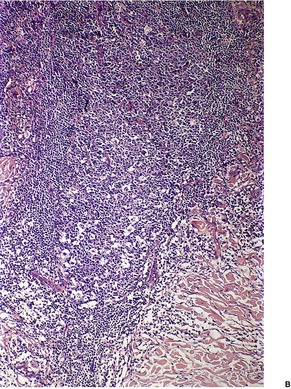 |
| Fig. 41.24 Cutaneous lymphoid hyperplasia. (A) The infiltrate is ‘top heavy’ and contains reactive follicles. (B) The germinal centers of reactive follicles contain tingible body macrophages. (H & E) |
In some comparative studies, reactive follicles were seen more often in cutaneous marginal zone lymphoma than in reactive lymphoid hyperplasia.1070. and 1072. Between these structures, there is an infiltrate rich in small T lymphocytes with admixed scattered T and B immunoblasts. Eosinophils, histiocytes, and plasma cells may also be present. Granulomas and necrosis can be seen in post-vaccination hyperplasia. 1056
Immunohistochemistry and cytogenetics
The B cells, T cells, and histiocytes mark with the appropriate markers for their lineage. The germinal centers stain for CD10 and are negative for bcl-2. Plasma cells are polyclonal when stained for immunoglobulin light chains. Dendritic cells positive for CD1a and S100 are also present in the infiltrate. 1067 Preliminary data suggest that the evaluation of B-cell clonality using the BIOMED-2 PCR system may be useful in distinguishing true cutaneous lymphomas from simulants. 1073
LYMPHOMATOID DRUG REACTIONS
The atypical lymphoid infiltrates in some cutaneous drug reactions can resemble mycosis fungoides (the so-called ‘T-cell pattern of cutaneous lymphoid hyperplasia’). The lesions associated with ingestion of drugs may take the form of solitary plaques, nodules or multiple lesions with a widespread distribution. In addition, erythroderma simulating Sézary syndrome and a digitate dermatosis-like pattern have been reported. Numerous drugs have been implicated including phenytoin sodium (hydantoin), carbamazepine, griseofulvin, atenolol, cyclosporine, allopurinol, ACE inhibitors, antihistamines, mexiletine, lisinopril, and valsartan.584.1074.1075.1076.1077.1078.1079.1080. and 1081.
Drug reactions do not always occur immediately after commencing a drug but a short clinical history is more typical of a lymphomatoid drug eruption than true lymphoma. 1032 The lesions usually regress after withdrawal of the drug but in the case of the anticonvulsants, the lesion may persist for 12 months or more.1082.1083. and 1084.
Histopathology
There is an infiltrate in the dermis which may be band-like, resembling mycosis fungoides, or nodular. 1074 Lymphomatoid vascular, and interstitial granulomatous reactions resembling the granulomatous patterns of mycosis fungoides have been described.689. and 1085.
The infiltrate often contains lymphocytes with atypical cerebriform nuclei. There is usually a substantial histiocytic component, particularly in the nodular lesions. Eosinophils and plasma cells are usually sparse or absent although they may be more obvious in the nodular infiltrates. 1080
Epidermotropism may be observed, sometimes with Pautrier microabscess-like collections. 1079 Large CD30+ T cells have been reported in the dermal infiltrate in reactions to carbamazepine. 1086
In the lymphomatoid vascular pattern, vessel walls are infiltrated by small and large lymphocytes and eosinophils. 1085
Immunohistochemistry and cytogenetics
The infiltrate consists predominantly of T cells, but rarely B cells predominate. This occurs with some antihistamines1080 and thioridazine. 1087 Most T cells are CD4+ and there is usually no loss of pan T-cell markers (CD2, CD3, CD5). 1088 Loss of expression of CD7 and CD62K has been described in some cases. 1085
REACTIONS RESEMBLING CD30+ LYMPHOPROLIFERATIVE DISORDERS
Atypical histological infiltrates with prominent large CD30+ cells can occur in a variety of situations1031 including drug eruptions (carbamazepine, gemcitabine, methotrexate),1053.1086. and 1091. leukemic infiltrates, 1031 the atypical eruption of lymphocyte recovery, 1092 viral infections including molluscum contagiosum (Fig. 41.25), herpes simplex, 1093 and lymphoproliferative lesions associated with EBV, 1094 arthropod bites including scabies1095 and tick bites, 1043 and gold acupuncture. 1096
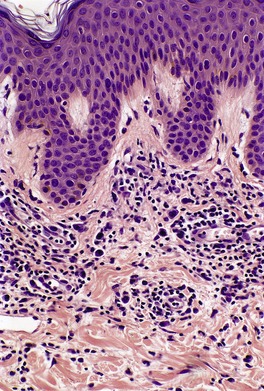 |
| Fig. 41.25 Atypical inflammatory cell infiltrate adjacent to molluscum contagiosum. The large cells are CD30-positive T cells. (H & E) |
PSEUDOLYMPHOMATOUS FOLLICULITIS
Pseudolymphomatous folliculitis is a condition which may mimic a cutaneous lymphoma clinically, and folliculotropic mycosis fungoides histologically. 246 Lesions occur on the face and are solitary, flat or dome-shaped erythematous papules or nodules up to 1.5 cm in diameter. Regression occurs in some cases after biopsy.
Histopathology
There is a diffuse, predominantly perifollicular infiltrate of lymphocytes with infiltration of follicular structures together with destruction of follicles or hyperplasia of follicular epithelium. Aggregates of histiocytes, or granulomas related to disrupted follicles are often seen; lymphoid follicles are rarely present. The epidermis is not usually involved. Large atypical lymphocytes and plasma cells can be seen in the infiltrates.246. and 1097.
Immunohistochemistry and cytogenetics
There is a mixture of B and T cells identifiable by conventional markers (CD20, CD79a, CD3, CD45RO). Either B or T cells may predominate. Plasma cells are polyclonal. In one case CD8+ cells predominated over CD4+ T cells in the follicular epithelium. Aggregates of perifollicular dendritic cells which are CD1a+ and S100+ are characteristic. Cells are negative for EBER-1, and neither B- nor T-cell clonality has been identified by PCR.246. and 1097.
JESSNER’S LYMPHOCYTIC INFILTRATE
Jessner’s lymphocytic infiltrate is a relatively uncommon condition of unknown etiology that was first delineated by Jessner and Kanof in 1953. 1098 It is not regarded by all as a distinct entity1099 and some consider it part of the spectrum of lupus erythematosus or polymorphic light eruption (see p. 57).1100. and 1101. It is discussed here as it has been classified as ‘pseudolymphoma’, 1074 although the histology is often not atypical and does not suggest a malignant process. In some cases it may histologically mimic the infiltrates of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL).
In this condition there are asymptomatic erythematous plaques, usually on the face or neck. 1102 The upper part of the trunk and other sites are occasionally involved. 1037 Men are predominantly affected, but it has been reported in children and in a familial setting. There is a recent report of a case following the use of a hydroquinone-containing bleaching cream. 1103 It has a benign but somewhat unpredictable course. Individual lesions may show central clearing and even regression, but there may be recurrences in the same or other areas. The average duration of the disease is 5 years.
Histopathology
There is a moderately dense perivascular infiltrate involving the superficial and deep vascular plexus as well as infiltrates surrounding pilosebaceous units; the subcutis may be involved. The component cells are predominantly small lymphocytes although larger lymphocytes and plasma cells may be present.1104.1105.1106.1107.1108.1109. and 1110. Mucin may be seen between collagen bundles.
The epidermis is usually normal with no evidence of atrophy, basal vacuolar change, or follicular plugging. These features together with negative immunofluorescence are said to distinguish this condition from lupus erythematosus. In 10–20% of cases (or more), lupus immunofluorescence is negative. 1100
Immunohistochemistry
There is predominantly a T-cell population with a smaller component of B cells. Clear distinction between cutaneous lupus and Jessner’s lymphocytic infiltrate cannot be made based on relative proportions of B and T cells in lesions. 1111 The cells of CLL are B cells which are CD20+, CD5+.
ACRAL PSEUDOLYMPHOMATOUS ANGIOKERATOMA
This condition was first characterized by Ramsay et al1112, who gave it the acronym ‘APACHE’ – acral pseudolymphomatous angiokeratoma of children. A more appropriate name would appear to be ‘papular angiolymphoid hyperplasia’1113 as it does not appear to be an angiokeratoma histologically, it occurs in adults as well as children, and it is not always acral in distribution. Clinically, it occurs most commonly in children between the ages of 2 and 13, 1112 but it has also been described in late adolescence and in adults.1114.1115. and 1116. Lesions are red in color and may take the form of single or multiple red papules which may have a linear configuration. 1117 Most lesions have been reported on the extremities but they have also been described at other sites.1114.1118. and 1119.
Histopathology
There is a confluent upper dermal infiltrate composed of small lymphocytes, histiocytes, and smaller numbers of plasma cells and eosinophils. Occasional giant cells may be seen. Within the infiltrate there is a proliferation of small vessels lined by plump endothelial cells. The overlying epidermis is thinned and there is a basal lichenoid reaction with apoptotic bodies or vacuolar change and exocytosis of lymphocytes.1114.1115.1117. and 1120. Lymphoid follicles have been demonstrated in a linear case. 1117
Immunohistochemistry and cytogenetics
The dermal infiltrate consists of a mix of T and B lymphocytes with characteristic markers: T cells predominate. The T cells are a mix of CD4+ and CD8+ cells and the epidermal cells are of similar lineage. In most cases PCR for TCR and IgH have not demonstrated clonality of T or B cells, 1116 except in one report in which a clonal IgH chain rearrangement was demonstrated. 1121
CUTANEOUS CD8+ T-CELL INFILTRATES IN HIV/AIDS
Cutaneous CD8+ T-cell infiltrates in HIV/AIDS present as plaques and nodules, predominantly on the face and extremities, or with generalized erythroderma.1122. and 1123. A variant, palpable arciform migratory erythema has also been described. 1124 Regression may occur after HIV antiviral triple therapy. 1125
Histopathology1122
In these cases there is a papillary and mid-dermal infiltrate of small cerebriform lymphocytes or a mixed population of lymphocytes including larger cells. Eosinophils are common in the infiltrate. There is mild epidermotropism and occasionally Pautrier microabscesses. Epidermal changes such as acanthosis and hyperkeratosis may also be present as well as a lichenoid reaction with vacuolar change and apoptotic bodies at the dermoepidermal junction. Papillary dermal fibrosis and granulomas are other changes.
Immunohistochemistry and cytogenetics
The cellular infiltrate consists predominantly of T cells which are CD2+, CD3+, CD5+, and CD8+. CD7 and CD4 are expressed by some cells. Clonal TCR gene rearrangements are not present.
MISCELLANEOUS
Several topics will be considered in this section. There are some similarities between these entities, although extramedullary hematopoiesis is completely different.
CUTANEOUS INFILTRATES IN POST-TRANSPLANT LYMPHOPROLIFERATIVE DISORDERS (PTLDs)
There is a heterogeneous group of lymphoproliferative disorders, usually of B-cell origin, which occur in the setting of organ transplantation (solid organ or bone marrow) and immunosuppression, particularly with cyclosporin A (cyclosporine) and monoclonal antibody OKT3. The risk of developing this complication overall was 1.2% in a large mixed series of transplants. 1126 It depends on the transplanted organ, the age at transplantation, the immunosuppressive drug regimen, 1127 and the EBV status of the patient prior to transplantation. 1128 The risk is approximately 1% for renal transplants, just over 2% for heart, lung and pancreas transplants, and 4.3 % for liver transplants. The risk is 2–3-fold higher in children. 1128 Approximately 80% of PTLDs show evidence of Epstein–Barr virus (EBV) infection.1128.1129. and 1130. Cutaneous lesions in the setting of PTLDs are very uncommon. Of those that are classified as lymphoma, 70% are B-cell and 30% T-cell types. 943 They are more common in men. Lymphomas occur most commonly after the first year of transplantation. 1127 Cutaneous lymphomas occur later after transplantation than non-cutaneous lymphomas. 943 The prognosis is better for B-cell lymphomas than T-cell lymphomas and is generally worse than that of the corresponding lymphoma in a non-transplant setting. 1131
The current WHO classification of PTLDs1132 is:
• Early lesions: Reactive plasmacytic hyperplasia and infectious mononucleosis-like
• Polymorphic PTLD
• Monomorphic PTLD (classified according to lymphoma type): diffuse large B-cell lymphoma; Burkitt/Burkitt-like lymphoma; plasma cell myeloma; plasmacytoma-like lesions
• T-cell neoplasms: peripheral T-cell lymphoma, not otherwise specified; other types
• Hodgkin lymphoma and Hodgkin lymphoma-like PTLD.
The early lesions occur most commonly in children and adults who are EBV-naive at time of transplantation. Presentation is within the first year after transplantation and there may be an infectious mononucleosis-like presentation. Immunoblasts in nodal infiltrates are EBV+ by immunohistochemistry or in-situ hybridization. The proliferation is usually polyclonal or oligoclonal. In most cases there is regression with reduction in immunosuppression. They have a good prognosis. Cutaneous lesions have not been reported in this form.
Related posts:
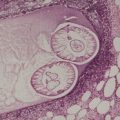
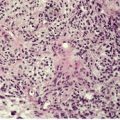
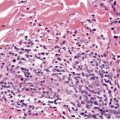
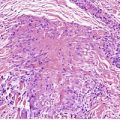
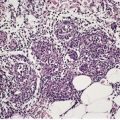
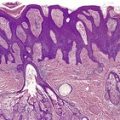
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


