Tumors of smooth muscle858
TUMORS OF SMOOTH MUSCLE
LEIOMYOMA
Histopathology
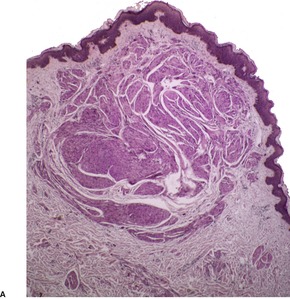
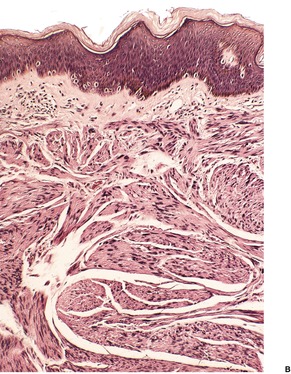
Fig. 36.1
CUTANEOUS AND UTERINE LEIOMYOMAS
Histopathology
ANGIOLEIOMYOMA
Histopathology
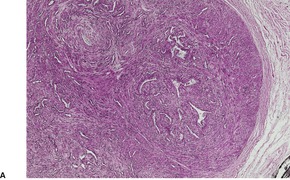
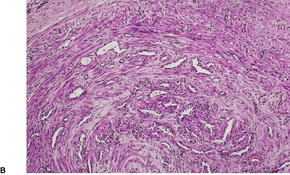
Fig. 36.2
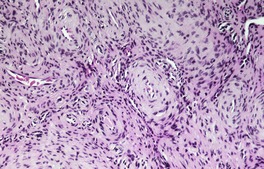
Fig. 36.3
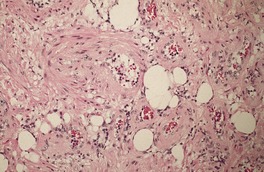
Fig. 36.4
SMOOTH MUSCLE HAMARTOMA
Histopathology
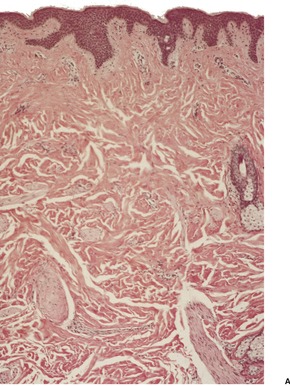
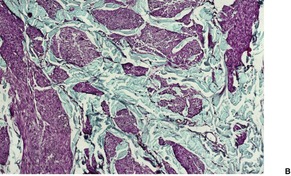
Fig. 36.5
LEIOMYOSARCOMA
Histopathology
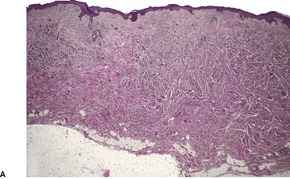
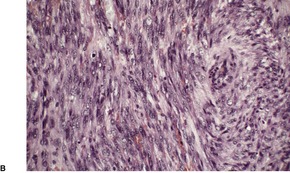
Fig. 36.6
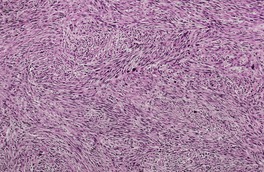
Fig. 36.7
Electron microscopy
TUMORS OF STRIATED MUSCLE
RHABDOMYOMATOUS MESENCHYMAL HAMARTOMA
Histopathology
RHABDOMYOMA
Histopathology
RHABDOMYOSARCOMA
Histopathology
MALIGNANT RHABDOID TUMOR
Histopathology
TUMORS OF CARTILAGE
CHONDROMA
Histopathology
SUBUNGUAL OSTEOCHONDROMA
Histopathology
PARACHORDOMA
Histopathology
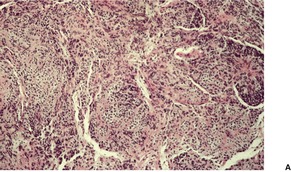
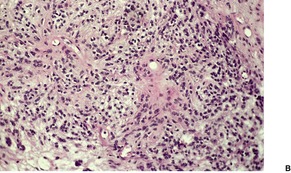
Fig. 36.8
TUMORS OF BONE
OSTEOSARCOMA
Histopathology
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Tumors of muscle, cartilage, and bone
Leiomyoma858
Cutaneous and uterine leiomyomas858
Angioleiomyoma859
Smooth muscle hamartoma860
Leiomyosarcoma861
Smooth muscle is found in the skin in three distinct settings: the arrector pili muscles, the walls of blood vessels, and the specialized muscle of genital skin, which includes the scrotum (dartos muscle), vulva and nipple (areolar smooth muscle). Each of these sources of smooth muscle can give rise to benign tumors, resulting in three categories of cutaneous leiomyoma: piloleiomyoma, leiomyoma of genital skin, and angioleiomyoma.1.2. and 3. The rare leiomyoma of deep soft tissue, and the case reported as ‘leiomyomatosis’ on the basis of numerous large and widespread tumoral masses of smooth muscle, are additional categories.4. and 5. Leiomyomas of the scrotum and vulva show some histopathological differences from piloleiomyomas and leiomyomas of the nipple. These differences will be highlighted below.
For completeness, it should be noted that smooth muscle has also been reported, rarely, in organoid6 and blue nevi. 7 It has been reported as the only stromal component in a folliculosebaceous cystic hamartoma. 8
Leiomyomas derived from the arrector pili muscle (piloleiomyomas, pilar leiomyomas) are more often multiple than solitary. Multiple lesions usually have their onset in the late second or third decade of life. They present as multiple, firm, reddish-brown papulonodules with a predilection for the face, 9 back, and extensor surfaces of the extremities. 10 Several hundred lesions may be present. They may cluster to form plaques,11.12. and 13. which usually involve more than one area of the body.14. and 15. Rarely, tumors are zosteriform or symmetrically distributed,16. and 17. suggesting a nevoid condition, and these cases have been designated ‘nevus leiomyomatosus systematicus’. 18 Some of these cases probably represent a type 2 segmental manifestation of cutaneous leiomyomatosis. 19 Some of the multiple cases are familial, with an autosomal dominant inheritance.20. and 21. There is a report of identical twins being involved. 22 Multiple leiomyomas have been associated with uterine leiomyomas in some females (see below). Multiple piloleiomyomas may occur on the breast, away from the nipple. It has been suggested that mechanical stretching in large pendulous breasts may play an etiological role. 23 Erythropoietic activity of the tumors is a rare finding. 24 Minor trauma or exposure to cold temperatures may lead to severe pain in the tumors.11.25. and 26.
Solitary piloleiomyomas,27. and 28. which are infrequently painful, are usually slightly larger than those found in patients with the multiple form, sometimes reaching 2 cm or more in diameter. Rarely they are present at birth.29. and 30. There has been a female preponderance.
There are only sparse reports of leiomyoma of the nipple14. and 31. in the dermatopathological literature. Multiple lesions on both nipples are extremely rare. 32 Leiomyomas of genital skin are quite uncommon, with only small numbers of scrotal and vulval lesions reported.14.31.33.34. and 35. Scrotal leiomyomas present as firm, solitary asymptomatic nodules, measuring 1–14 cm in diameter. 36Vulval leiomyomas usually arise in the labia majora.31. and 35. They usually measure 1–5 cm in diameter. Most are asymptomatic.
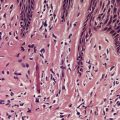
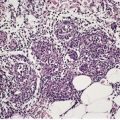
Piloleiomyomas are circumscribed non-encapsulated tumors, centered on the dermis.14. and 15. An overlying zone of uninvolved subepidermal tissue (so-called ‘grenz zone’) is usually present, and there may be some flattening of the epidermis. Epidermal hyperplasia was present in over 50% of cases in one series. 2 The tumor is composed of bundles of smooth muscle arranged in an interlacing and sometimes a whorled pattern (Fig. 36.1). The cells have abundant eosinophilic cytoplasm and elongated nuclei with blunt ends. Nuclear palisading and the formation of Verocay bodies was reported in one case. 37 Granular cell change is a rare variant. 38 There are usually no mitoses in the pilar variant. Tumors of long standing may have fibrous tissue in the stroma, and occasionally this shows focal hyalinization. 31 Osseous metaplasia is rare. 39 Focal stromal myxoid change and small lymphoid collections have been noted. Hair follicles are sometimes found surviving within the tumor. 14 The smooth muscle nature of the cells can be confirmed with the Masson trichrome stain or the use of immunoperoxidase markers for smooth muscle such as vimentin, smooth muscle actin (SMA), desmin, and caldesmon. 39 SMA is more specific for smooth muscle than panactin HHF35. 40 Lesions on the nipple are estrogen and progesterone receptor positive; both receptors are also found in normal subareolar muscle. 41 A Bodian stain has shown increased numbers of nerve fibers interlacing with the muscle fibers, and also in the surrounding tissue. 15
(A) Leiomyoma. (B) The reticular dermis is replaced by interlacing bundles of smooth muscle cells. (H & E)
Scrotal leiomyomas often have ill-defined or focally infiltrative margins, differing from the circumscribed tumors of pilar origin. Scrotal lesions are often more cellular with occasional mitoses. Symplastic features have been reported in all types.31.42.43. and 44. Clear cell and granular cell variants are rare. 45 Lymphoid aggregates are sometimes present. Dartoic muscle is seen adjacent to the tumor. Male genital leiomyomas may express androgen receptors. 46
Vulval leiomyomas are usually of spindle-cell type, although rare epithelioid and myxoid variants occur.31. and 47. Sparse mitoses may be present. Stromal hyalinization is found in nearly one-half of the cases. Small fascicles of spindle cells may be trapped in these hyaline areas producing a plexiform appearance. 31 The majority of cases express estrogen and progesterone receptors, 47 a feature not found in pilar lesions. 48
Smooth muscle hamartoma differs from leiomyoma by having discrete bundles of smooth muscle fibers set in dermal collagen (see below). The multiple papular variant of pilar leiomyoma not infrequently exhibits small bundles of collagen between the smooth muscle bundles, but usually the collagen is not as plentiful as it is in smooth muscle hamartoma. 49
The syndrome of multiple cutaneous and uterine leiomyomas (Reed syndrome, leiomyomatosis cutis et uteri – OMIM 150800) is a rare autosomal dominant disorder resulting from a germline mutation in the fumarate hydratase (FH) gene, which maps to chromosome 1q42.3–q43.24.50.51.52.53. and 54. Fumarate hydratase is an enzyme that functions as part of the Krebs cycle; it converts fumarate to malate.55. and 56. The gene may also have tumor suppressor functions. Many distinct mutations have been reported. 57 This syndrome can also be associated with type II papillary renal cell carcinoma and renal collecting duct carcinoma.54.55.56.57.58. and 59. Other morphological patterns have been described. 60 Leiomyosarcoma of the uterus is a rare complication. 57
The cutaneous lesions usually present in the second and third decades. They usually precede the diagnosis of uterine leiomyomas, which are often painful. 61 Nearly all women with multiple cutaneous leiomyomas have early-onset uterine leiomyomas. 62 The G354R mutation predisposes patients to uterine leiomyomas without skin leiomyomas. 61 Males with FH mutations also have cutaneous leiomyomas. 57
All cutaneous leiomyomas in this syndrome are of pilar origin. They are localized to the upper two-thirds of the reticular dermis. Some are well circumscribed and nodular, but most are diffuse in arrangement.
Angioleiomyoma usually presents as a solitary, slow-growing nodule on the extremities, particularly the lower leg, of middle-aged individuals.63. and 64. Digital lesions are rare.65. and 66. There is a female preponderance.67. and 68. More than half the lesions are painful or tender, but this feature is usually absent in those found on the face and upper trunk.63.69. and 70.
Multiple subcutaneous angioleiomyomas have been reported in a patient with acquired immunodeficiency syndrome (AIDS). 71 Epstein–Barr virus was demonstrated in the nuclei of the smooth muscle cells. 71
The tumors are firm, gray-white round to oval nodules in the lower dermis and subcutis. 72 They are usually less than 2 cm in diameter. Most authors assume that they arise from veins, although some may be hamartomas. 68 A rare intravascular variant has been reported. 73
Surgical excision is curative.
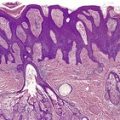
The tumor is usually well circumscribed with a fibrous capsule of variable thickness and completeness (Fig. 36.2). The main component is smooth muscle, which is present as interlacing bundles between the numerous vascular channels.63.68. and 72. Most of the vessels have several layers of smooth muscle in the walls which often merges peripherally with the intervascular fascicles (Fig. 36.3). Sometimes there are large sinusoidal vessels with little smooth muscle in their walls. Small slit-like channels are sometimes present. Most vessels have only scant and scattered elastic fibers in their walls.
(A) Angioleiomyoma. (B) The smooth muscle bundles are admixed with small blood vessels. (H & E)
Angioleiomyoma. In this case the individual vessels are thick walled; their outer layers of smooth muscle merge with the intervascular muscle fascicles. (H & E)
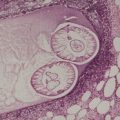
The stroma contains varying amounts of fibrous tissue, and in about a third of cases there is a sparse lymphocytic infiltrate. Myxoid change is quite common in the stroma, particularly in the larger tumors. 67 The presence of fat in a few cases has led to suggestions of a hamartomatous origin; 68 a designation of ‘angiomyolipoma’ can be used for these cases which also contain fat (Fig. 36.4).74.75.76.77.78. and 79. The HMB-45 is negative in cutaneous tumors, in contrast to the finding in renal lesions.80. and 81. For these reasons, cutaneous angiomyolipomas should be regarded as an angioleiomyoma with fat.82. and 83.The term ‘myolipoma’ is used for the rare tumors containing smooth muscle and adipose tissue, but no vascular component. 84 Uncommon changes include thrombosis of vascular channels, focal calcification, 85 stromal hemosiderin, and hyalinization of vessel walls. Nuclear palisading, producing Verocay-like bodies, is a rare occurrence. 86 Nerve fibers are only seen occasionally. 1 An angioleiomyoma arising in a histiocytoma has been reported. 87
Angiomyolipoma. There is an admixture of blood vessels, smooth muscle, and adipocytes. (H & E)
In a review of 562 cases, three histological variants were described: a solid type, in which smooth muscle bundles surround numerous small slit-like channels; a cavernous type, with dilated vascular channels, the walls of which are difficult to distinguish from the intervascular smooth muscle; and a venous type, with thick-walled vessels which are easily distinguished from the intervascular smooth muscle. 68 Using this classification, a more recent study has found that of 122 cases, 74 were of solid type, 11 were cavernous and 37 venous in type. 70 In this study, the authors commented on the close histological resemblance of cases with a concentric perivascular arrangement to myopericytoma, a finding noted earlier by Mentzel et al. 88 Another variant is the epithelioid type, which is composed of cells with round to oval nuclei and a moderate amount of finely granular eosinophilic cytoplasm with occasional vacuoles. 89 An epithelioid variant with clear cell change has also been reported. 90 The pleomorphic type has marked nuclear pleomorphism but only rare or absent mitoses. There is some resemblance to the symplastic change seen in uterine leiomyomas.91.92. and 93.
In the series of 122 cases (mentioned above), all cases were diffusely positive for actins (α-smooth muscle actin and HHF35) and calponin and diffusely or focally positive for h-caldesmon. 70 Desmin was diffusely positive in three-quarters of solid-type angioleiomyomas, a half of the venous type, but only 18% of the cavernous type. 70 In contrast most of the myopericytomas were negative for desmin, but they stained for the actins and calponin, and, with one exception, also h-caldesmon. 70
The smooth muscle hamartoma is a rare, usually congenital, hyperplasia of dermal smooth muscle fibers which presents as a flesh-colored or lightly pigmented plaque up to 10 cm in diameter on the extremities or trunk.3.94.95.96.97.98.99.100.101. and 102. The scrotum and prepuce are rarely involved.103.104.105. and 106. Some of the scrotal cases appear to represent acquired smooth muscle hypertrophy in response to chronic lymphedema. 107 Within the plaques small gooseflesh-like papules may be discernible, and these may transiently elevate when rubbed (pseudo-Darier’s sign).108.109. and 110. This phenomenon of spontaneous, often asymptomatic, undulations of muscle fibers is known as myokymia. It may be the presenting sign of congenital smooth muscle hamartoma. 111 A rare linear variant with perifollicular papules has been reported. 112 Hairs are usually more prominent in the skin of the affected site, being slightly longer and thicker than in the adjoining skin. 113 A more generalized variant with prominent skin folds has been reported as a ‘Michelin tire baby’.114.115.116.117. and 118. Follicular dimpling and hypertrichosis may be present in this generalized variant. 119 Multiple lesions have been described in three members of the same family. 120
Smooth muscle bundles, indistinguishable from those seen in this condition, may also occur in Becker’s nevus,121.122. and 123. and this has led to some controversy about the relationship of these two entities.108. and 113. Becker’s nevus has its onset in adolescence, with invariable hyperpigmentation and hypertrichosis. Recent reports of patients with the clinical features of smooth muscle hamartoma which developed in late childhood124 or early adulthood125.126. and 127. suggest that Becker’s nevus and smooth muscle hamartoma belong at different poles of the same developmental spectrum, involving hamartomatous change to the pilar unit and arrectores pilorum.100.128. and 129. A smooth muscle hamartoma has been reported in association with a melanocytic nevus; a Becker’s nevus has also been present in one case. 130
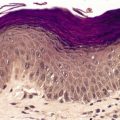
There are well-defined smooth muscle bundles in the dermis, oriented in various directions. Some are attached to, or surround, hair follicles.100. and 108. Often there is a thin retraction space around the bundles, separating them from the adjacent dermal collagen (Fig. 36.5). The smooth muscle bundles are clearly seen with the Masson trichrome stain. Bundles of nerve fibers may also be present. 99 These findings can be confirmed with appropriate immunohistochemical stains. The smooth muscle expresses desmin and smooth muscle actin.102.105. and 107. Interestingly, large numbers of CD34-positive cells may be present in the stroma surrounding the smooth muscle bundles. 131 They were not present in the acquired scrotal cases secondary to lymphedema. 107 There is often slight elongation of the rete ridges of the overlying epidermis, and mild basal hypermelanosis. 128
Smooth muscle hamartoma. (A) There are scattered smooth muscle bundles in the dermis. (H & E) (B) There is collagen between the smooth muscle bundles. (Masson trichrome)
The smooth muscle hamartoma reported in a patient with the MLS syndrome (microphthalmia with linear skin defects) appeared to consist of hypertrophied arrectores pilorum muscle with some thinning of the epidermis and upper dermis. 132
Primary cutaneous leiomyosarcomas are infrequent tumors that may arise in the dermis or subcutaneous tissue.133. and 134. Secondary leiomyosarcomas constitute a third category; they are exceedingly rare. Dermal and subcutaneous lesions have a different biological behavior (see below).
Well over 100 cases of dermal leiomyosarcoma have now been recorded.133. and 135. These tumors have a predilection for the extensor surfaces of the extremities,133. and 134. and to a lesser extent the scalp, 136 and trunk.137.138.139.140. and 141. Rare sites include the upper lip, 142 nipple,143. and 144. penis,145. and 146. scrotum,147. and 148. vulva, 149 a chronic venous stasis ulcer, 150 and the face.27. and 151. There is a male predominance, and the average age of presentation is in the sixth decade. Childhood presentation is rare.152. and 153. The tumors vary in size from 0.5 to 3 cm or more in maximum diameter. Subcutaneous extension is present in two-thirds of cases. 133 Pain or tenderness is present in some. 134 Occasionally there is a history of previous injury to the site.134. and 137. These tumors presumably arise from the arrector pili muscles, except for scrotal lesions, which derive from the dartos muscle.133. and 134. Cases arising in an angioleiomyoma, 154 in an organoid nevus (nevus sebaceous), 155 and in the scar of a previously excised leiomyoma have been reported. 156 Dermal leiomyosarcomas may recur locally in up to 30% of cases (10% in the author’s experience), but metastases of confirmed cases are unknown. 133
The recurrence rate seems to be lower when wide surgical excision is carried out initially. A clearance of at least 3 cm should be obtained. Mohs surgery has been used for lesions on the face. 157
Subcutaneous leiomyosarcomas tend to be slightly larger at presentation and more circumscribed in outline. 134 They may also be tender or painful. They presumably arise from the smooth muscle in vessel walls. 158 They have a greater tendency for local recurrence (50–70%), and metastases to lung, liver, bone, and other sites occur in about one-third of cases.134.159. and 160. Assessment of the DNA content of the tumor cells by flow cytometry may be used to predict those with metastatic potential.161.162. and 163. The most frequent genomic alterations in leiomyosarcomas involve losses in the 13q4–q21 region, but other chromosomal losses occur. 164 Malignant fibrous histiocytoma shares many of these genomic imbalances. 164
Secondary leiomyosarcomas are rare in the skin and arise from retroperitoneal and uterine primary lesions. 165 There are usually several dermal or subcutaneous nodules. There has been a predilection for the scalp and back.
Dermal leiomyosarcomas are irregular in outline, with tumor cells blending into the collagenous stroma at the periphery. 166 By definition, the major portion of the tumor is in the dermis, although subcutaneous extension occurs in two-thirds. 133 A superficial grenz zone is present in many. Ulceration and pseudoepitheliomatous hyperplasia are rare. There is usually some flattening of the rete ridges of the overlying epidermis.
Leiomyosarcomas are composed of interlacing fascicles of elongated spindle-shaped cells with eosinophilic cytoplasm and eccentric, blunt-ended (cigar-shaped) nuclei (Fig. 36.6). Rare variants with intracytoplasmic eosinophilic granules (granular cell leiomyosarcoma)38.167. and 168. or with epithelioid cells (epithelioid leiomyosarcoma)169. and 170. have been reported. Sometimes there is a suggestion of nuclear palisading. There is variable nuclear pleomorphism, with at least one mitosis per 10 high-power fields in cellular areas. Pockets of greater mitotic activity (mitotic ‘hot spots’) 171 are found. Tumor giant cells are usually present in the less well-differentiated variants. Perinuclear halos are rare. Small lymphoid aggregates are sometimes present within the tumor, 133 but a dense infiltrate (inflammatory leiomyosarcoma) is quite rare. 152 Stromal sclerosis (desmoplastic leiomyosarcoma) is a rare phenomenon.151.172.173. and 174. The resemblance of this tumor to other desmoplastic tumors of the skin is striking and immunohistochemistry is necessary for its diagnosis. 173 Arrector pili muscles are often prominent or hyperplastic, and in some cases transitions from normal to hyperplastic, to benign neoplastic, and to a frankly sarcomatous pattern occur.133. and 134.
Leiomyosarcoma. (A) The tumor is composed of plump spindle-shaped cells. (B) Scattered mitoses are present. (H & E)
In one large series, two predominant growth patterns were observed: nodular and diffuse (infiltrative). 135 Nodular tumors were usually quite cellular with nuclear atypia and many mitoses. Sometimes there were small foci of necrosis. Diffuse tumors were less cellular with well-differentiated smooth muscle cells and inconspicuous mitoses. 135
Subcutaneous leiomyosarcomas often extend into the lower dermis (Fig. 36.7). They frequently have a prominent vascular pattern; 134 vascular invasion is an adverse prognostic feature. 162 Small areas of necrosis are sometimes present. Deep variants with either a heavy inflammatory cell component175 or osteoclast-like giant cells176 have been reported. A myxoid and a pleomorphic variant have been reported in the deeper soft tissues.177.178.179. and 180.
Subcutaneous leiomyosarcoma. The lesion is quite cellular. (H & E)
Secondary leiomyosarcomas are often multiple, spheroidal in outline, and sometimes present in vascular lumina.133. and 181.
Leiomyosarcomas of all three types will have myofilaments, demonstrated by the Masson trichrome stain. Reticulin stains show a fine reticulin network interspersed between adjacent fibers. A small amount of glycogen is usually present. 182 Immunoperoxidase preparations show the presence of vimentin, smooth muscle actin, and h-caldesmon.174. and 183. Desmin is present in about 70% of cases but is less common in the higher-grade tumors.135. and 163. Pan-muscle actin (HHF35) is usually present. Cytokeratin, CD117, S100 protein, and factor XIIIa have been demonstrated in a small number of cases.135.184.185. and 186. In the case of the cytokeratin marker MNF116, Iwata and Fletcher found positivity in 5 of 20 cutaneous cases; EMA was expressed in nine of these cases. 187β-catenin is not expressed.
Reliable criteria for malignancy remain to be established. 171 Usually accepted features include high cellularity, significant nuclear atypia, tumor giant cells, and at least one mitosis per 10 high-power fields. Tumor size of 5 cm or more is an adverse feature in subcutaneous tumors. 188 The cell-matrix adhesion protein migfilin is increased in the cytoplasm of tumor cells in the progression of leiomyosarcomas from low to high grade. 189 It is not found in normal smooth muscle cells.
Electron microscopy confirms the smooth muscle origin of the tumor cells, with numerous fine myofilaments in the cytoplasm, some marginal plaques, pinocytotic vesicles, glycogen, and a basal lamina surrounding individual cells.147. and 171. Junctional complexes are uncommon. 171 Some myofibroblasts may also be present. 148
There is an exceedingly rare group of cutaneous tumors that contain either mature striated muscle, or cells differentiating toward striated muscle, as a major component of the lesion. Mature striated muscle is found in some parts of the face as a normal constituent, 190 in rhabdomyomatous mesenchymal hamartoma (see below), the accessory tragus (see p. 502), and in the first arch abnormality reported as dermatorynchus geneae (see p. 502). Rarely, a few striated muscle fibers may be present adjacent to developmental cysts of the head and neck. There is one report of skeletal muscle regeneration presenting as an enlarging nodule on the lip following blunt trauma. 191 Rhabdomyomas are usually found in the vicinity of the aerodigestive tract. 192 Rarely, they occur elsewhere (see below). Malignant cells showing ultrastructural, immunohistochemical, and sometimes light microscopic differentiation toward striated muscle are a feature of rhabdomyosarcomas. Brief mention will also be made of the malignant rhabdoid tumor, which has some histopathological similarities to a rhabdomyosarcoma, but which is of uncertain histogenesis.
This rare hamartoma of the deep dermis and subcutaneous fat was first described in 1986 as a striated muscle hamartoma. 193 It has also been reported as congenital midline hamartoma. 194 It may present as a solitary papule or nodule in a normal neonate or arise in the setting of multiple ectodermal/mesodermal abnormalities.195.196.197.198.199. and 200. Such cases are called multiple rhabdomyomatous mesenchymal hamartomas. 201 Solitary lesions are usually midline in location on the head and neck. The chin is a common site; 202 the perianal region is rarely involved.203. and 204.
The polypoid tumors contain multiple bundles of normal-appearing striated muscle surrounded by fibrofatty tissue containing a few telangiectatic vessels.193. and 196. Smooth muscle as well as striated muscle was present in the lesion from the perianal region reported as a cutaneous mesenchymal hamartoma with mixed myogenous differentiation. 204 Numerous vellus hair follicles or folliculosebaceous structures are usually present. 205 Lentiginous melanocytic hyperplasia was present in one case. 206 It was not known if this was a component of the hamartoma or an inductive phenomenon. 206 The worm-like, flesh-colored, cutaneous polyps that occur in the disorganization syndrome (OMIM 223200) lack striated muscle, a feature of multiple rhabdomyomatous mesenchymal hamartomas. 201
The subcutaneous tumor with desmin-positive spindle cells, reported as pleomorphic hamartoma of the subcutis, is of uncertain histogenesis. 207
Two variants of rhabdomyoma have been described: the adult rhabdomyoma and the fetal rhabdomyoma. 208 The adult rhabdomyoma is a rare, benign neoplasm that usually occurs in the head and neck region. 209 Rarely, it may arise elsewhere. 210 Rhabdomyomas are well-circumscribed, soft tumors with a reddish-brown appearance. They may recur locally if incompletely excised. The fetal rhabdomyoma is found in both children and adults. It is located in various organs of the body. Only one case involving the skin has been reported. 208 It may develop in deeper tissues in the nevoid basal cell carcinoma syndrome.
Adult rhabdomyomas are composed of sheets of large polygonal cells with granular, eosinophilic cytoplasm. The cells are partly vacuolated due to their glycogen content. ‘Spider-web’ cells, with residual strands of cytoplasm between the vacuoles, and cells with cross-striations may both be present. 209 The tumor cells stain strongly for desmin and myoglobin; some may stain weakly for S100 protein. 210
Fetal rhabdomyomas are composed of immature striated muscle. Fascicular bundles of immature cells with oval to spindle-shaped nuclei are present. 208 Scattered among these bundles are elongated cells with rhabdomyoblastic differentiation.
Rhabdomyosarcoma, a tumor composed of malignant cells showing some differentiation toward striated muscle, has a predilection for the head and neck region,211. and 212. the genitourinary tract, the retroperitoneum and the soft tissues of the extremities. It is the most common soft tissue sarcoma of childhood. Rarely, rhabdomyosarcoma may present as a dermal nodule, particularly on the head or neck, usually as a result of the dermal extension of a lesion arising in the underlying soft tissues.213.214.215. and 216. Lesions of dermal origin are rare.217. and 218. A primary cutaneous presentation occurs in less than 1% of all rhabdomyosarcomas. 219
Rhabdomyosarcomas have been reported in patients with neurofibromatosis, in the basal cell nevus syndrome, and many years after radiotherapy. 220
Specific chromosomal translocations involving chromosome 13 are consistently found in the alveolar subtype; chromosomal gains and losses have been reported in the embryonal form but not any diagnostic translocations.214. and 219.
There are three major histological subtypes of rhabdomyosarcoma: embryonal (including the botryoid variant), alveolar, 221 and pleomorphic.222. and 223. The spindle-cell rhabdomyosarcoma is a rare variant of the embryonal type. 216 Rhabdomyosarcomas may be composed of small round cells, spindle or polygonal cells, or large pleomorphic cells. Cross-striations are sometimes seen using the phosphotungstic-acid–hematoxylin stain.
Immunoperoxidase techniques using monoclonal antibodies to vimentin, desmin, myogenin, myoglobin, MyoD1, Myf-4, and muscle-specific actin may be used to confirm the diagnosis.213.216. and 218. Myogenin and MyoD1 are very specific for striated muscle, but they may be negative in some pleomorphic rhabdomyosarcomas. 40 In one cutaneous embryonal rhabdomyosarcoma, the spindle cells stained for S100 protein. 215 Some tumors, particularly the alveolar variant, express ALK protein. 224
Malignant rhabdoid tumors (OMIM 609322) were initially regarded as a subset of Wilms’ tumor of the kidney showing rhabdomyosarcomatous differentiation. 225 Subsequent studies have not confirmed this, and their histogenesis is currently uncertain. Tumors of similar morphology to the renal lesions have been reported in the skin.226.227.228.229.230.231. and 232. Cutaneous lesions are highly aggressive neoplasms, often present at birth, although adult cases have been reported. 233 Sometimes this rhabdoid phenotype is seen in conjunction with other cutaneous tumors. 233 The average survival following diagnosis is less than 6 weeks. 234 Genetic studies show a consistent pattern of abnormalities of chromosome 22q11, involving deletions and/or translocations.234. and 235. The affected gene is INI1, also known as SMARCB1. 236 Familial cases involving multiple generations have resulted from mutations in this gene. 236
There is a report of two children with papulonodular skin lesions, present at birth, who subsequently developed malignant rhabdoid tumors, in one case in contiguity. The term ‘neurovascular hamartoma’ was used for these lesions composed of bland spindle cells in a vascular stroma. 237 Another case developed adjacent to a ‘benign myofibromatous proliferation’. 238
The tumor is composed of sheets of polygonal cells with abundant hyaline eosinophilic cytoplasm and a peripherally displaced vesicular nucleus. Mitotic activity is prominent, and there are usually areas of tumor necrosis. 226
The cells invariably express vimentin and cytokeratins AE1/AE3 and/or CAM5.2. They often show strong focal to diffuse membranous staining for epithelial membrane antigen.234. and 239. Focal positivity for muscle-specific actin and desmin may occur. 234 Malignant rhabdoid tumor has some morphological and immunohistochemical features in common with epithelioid sarcoma (see p. 840), although the cells in epithelioid sarcoma do not contain rhabdoid filamentous masses.226. and 227. Rhabdoid differentiation has been reported in some malignant nerve sheath tumors. 240
Cartilage can be found in the skin in hamartomas (see p. 375), in certain tumors (such as chondroid syringomas), and in chondromas and osteochondromas. The latter two entities will be discussed here, together with the morphologically similar tumor known as parachordoma. The extraskeletal myxoid chondrosarcoma is a deep soft tissue tumor and beyond the scope of this book. 241 A chondroblastoma, presenting as a subungual mass on a toe, has also been reported. 242
True cutaneous chondromas, without bony connection, are exceedingly rare.243. and 244. They occur most commonly on the fingers, but have been recorded at other sites, such as the ear245 and nose. 245 Multiple lesions were present in one patient, who had a suggested autosomal dominant mode of inheritance. 245
Chondroma is a well-circumscribed expansile tumor composed of single and grouped chondrocytes embedded in a cartilaginous matrix. It usually produces effacement of the epidermal rete ridge pattern. 245 The cells exhibit reactivity for S100 protein.
Subungual osteochondroma (exostosis) is a fairly common, often painful, tumor occurring on the distal phalanx of a digit, usually the great toe.246.247.248.249.250. and 251. They may be misdiagnosed initially. 252
Excision of the lesion is the usual treatment. 253
The lesion consists of a base of mature trabecular bone with a proliferating cap of mature cartilage. 250 Osteogenesis occurs by endochondral ossification.
Parachordoma is an exceedingly rare soft tissue neoplasm with a superficial resemblance to a chordoma. It develops on the extremities adjacent to a tendon, synovium, or bone, unlike chordoma which is axial in location.254. and 255. Parachordomas are slow-growing tumors with occasional late recurrence. Cases with reported metastasis have not been convincing. 256
Chordomas may develop in the skin by direct extension from an underlying sacrococcygeal tumor, or by distant metastasis (see p. 936). The term chordoma cutis has been used for such cases. 257
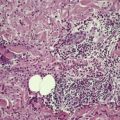
As in chordomas, there are strands and cords of vacuolated (physalipherous) cells set in a myxoid stroma (Fig. 36.8). 254 The cells stain for S100 protein, vimentin and epithelial membrane antigen.255. and 256. Parachordomas are strongly positive for cytokeratin 8/18, but not for CK1/10, 7, and 20. 256 Extraskeletal myxoid chondrosarcoma is negative for all cytokeratins.256. and 258.
(A) Parachordoma. (B) Nests of vacuolated cells are set in a fibromyxoid stroma. (H & E)
Mature bone may be found in the skin in the various forms of osteoma cutis (see p. 372). True neoplasms with differentiation toward bone (osteosarcoma) are exceedingly rare in the skin.259.260. and 261. A case purporting to be a cutaneous osteoblastoma has been reported. 262
Osteosarcoma (osteogenic sarcoma) of the skin may arise de novo or within heterotopic bone.259.263.264. and 265. An origin from underlying bone or metastasis from a primary lesion elsewhere must be excluded. Primary osteosarcoma of the skin can recur locally261 and/or spread to the lungs and other organs.259.260. and 266.
The tumor may be found in the dermis and/or the subcutis. 267 Ulceration often occurs. Highly cellular areas composed of spindle cells with scant eosinophilic cytoplasm and elongated hyperchromatic nuclei are usually present, in addition to foci of chondroid and osteoid differentiation. 265 The tumor cells are positive for vimentin but negative for S100 protein and cytokeratins.
References
Tumors of smooth muscle
1. Montgomery, H; Winkelmann, RK, Smooth-muscle tumors of the skin, Arch Dermatol 79 (1959) 32–39.