Introduction248
INTRODUCTION
Structure of the epidermis
Keratinization
Readers should refer to the new consensus nomenclature for mammalian keratins published by Schweizer et al in J Cell Biol 2006; 174: 169–174.
Keratin type
Primary location
K1, K10
Suprabasal cells
K1b (now K77)
Eccrine sweat glands
K2e
Late suprabasal cells
K2p (now K76)
Hard palate
K3, K12
Cornea
K4, K13
Mucosa
K5, K14
Basal keratinocytes
K6a, K16
Palmoplantar, mucosa, appendages
K7
Myoepithelia, simple epithelia
K8, K18
Simple epithelia
K9
Palmoplantar
K11
Polymorphic variant of K10
K15
Basal keratinocytes, outer root sheath
K17, K6b
Epidermal appendages
K19
Simple epithelia, epidermal appendages
K20
Gastrointestinal tract
hHb5 (now K85)
Hair, nails
hHb6, hHb1 (now K81, K86)
Cortical trichocytes
K25irs 1–4
Inner root sheath (type 1 keratins)
K71–74
Inner root sheath (type II keratins)
Disease
Keratin mutation
Bullous congenital ichthyosiform erythroderma
K1, K10
Ichthyosis bullosa of Siemens
K2e
Ichthyosis hystrix of Curth–Macklin
K1
Epidermolytic palmoplantar keratoderma
K9, K1 (some cases)
Non-epidermolytic palmoplantar keratoderma
K1 (one family) type II keratin cluster (not yet clarified)
Palmoplantar keratosis with anogenital leukokeratosis
K6, K16
Focal non-epidermolytic palmoplantar keratoderma
K1, K16
Unna–Thost palmoplantar keratoderma
K1, K16
Unilateral palmoplantar verrucous nevus
K16
Striate palmoplantar keratoderma (type III)
K1
Pachyonychia congenita
K6a, K6b, K16, K17
White sponge nevus
K4, K13
Monilethrix
K81, K86
Epidermolysis bullosa simplex
K5, K14
Abnormal process
Diseases with this abnormal process
Calcium pump
Darier’s disease
Hailey–Hailey disease
Sulfatase disorders
X-linked ichthyosis
Kallmann’s syndrome (ichthyosis and hypogonadism)
Multiple sulfatase deficiency
X-linked recessive chondrodysplasia punctata
Keratin disorders
See Table 9.2
Desmosomal defects
(desmoplakin; cadherins)
Carvajal syndrome (desmoplakin)
Striate palmoplantar keratoderma (type I desmoglein; II desmoplakin)
Naxos disease (plakoglobin)
Ectodermal dysplasia/skin fragility syndrome (plakophilin)
Tight junction (claudin)
ILVASC
Processing of filaggrin
Granular parakeratosis
Ichthyosis vulgaris (AGL variant)
Loricrin
Progressive symmetrical erythrokeratodermia (PSEK)-loricrin variant
Vohwinkel’s syndrome (ichthyotic variant)
Precursor proteins of cell envelope
Camisa keratoderma
PSEK (loricrin variant)
Absence of LEKT1
Netherton’s syndrome
Connexin
Erythrokeratodermia variabilis
Vohwinkel’s syndrome (keratoderma variant)
KID syndrome
Hidrotic ectodermal dysplasia
Oculodentodigital dysplasia
Abnormal lipid metabolism
Sjögren–Larsson syndrome (fatty aldehyde dehydrogenase gene)
Conradi–Hünermann–Happle syndrome (included as peroxisomal by some)
CHILD syndrome (included as peroxisomal by some)
Neutral lipid storage disease
Peroxisomes
Refsum’s disease (increased phytanic acid)
Chondrodysplasia punctata (two variants)
Lamellar granules
Harlequin ichthyosis
Lamellar ichthyosis
Congenital ichthyosiform erythroderma (50%)
Peeling skin syndrome (acral type)
Proteinase disorders
Papillon–Lefèvre syndrome (cathepsin C gene)
Haim–Munk syndrome
ICHTHYOSES
ICHTHYOSIS VULGARIS
Histopathology
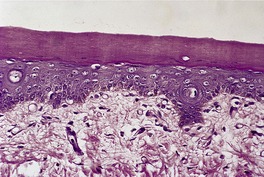
Fig. 9.1
Electron microscopy
X-LINKED ICHTHYOSIS
Histopathology
ICHTHYOSIS CONGENITA
Type I
Type II
Type III
Lamellar ichthyosis
Congenital ichthyosiform erythroderma
Histopathology
Fig. 9.2
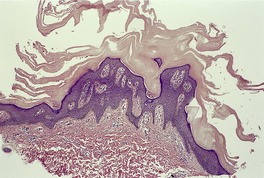
Fig. 9.3
Electron microscopy
BULLOUS ICHTHYOSIS
Histopathology
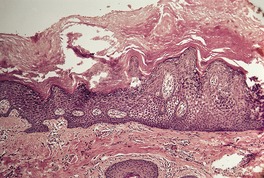
Fig. 9.4
Electron microscopy
NETHERTON’S SYNDROME
Histopathology
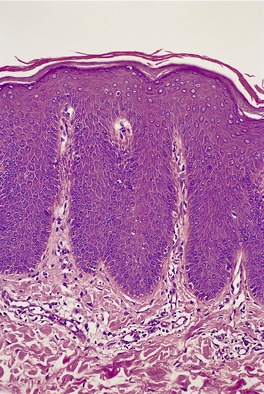
Fig. 9.5
ERYTHROKERATODERMIA VARIABILIS
Histopathology
HARLEQUIN ICHTHYOSIS
Histopathology
Electron microscopy281
FOLLICULAR ICHTHYOSIS
Histopathology
ACQUIRED ICHTHYOSIS
REFSUM’S DISEASE
Histopathology
Electron microscopy
OTHER ICHTHYOSIS-RELATED SYNDROMES
Sjögren–Larsson syndrome
‘KID’ syndrome
Conradi–Hünermann–Happle syndrome
Neu–Laxova syndrome
‘CHILD’ syndrome
‘IBIDS’ (ichthyosis with trichothiodystrophy)
‘ILVASC’
Multiple sulfatase deficiency
‘MAUIE’ syndrome
Neutral lipid storage disease (Dorfman–Chanarin syndrome)
Shwachman syndrome
PALMOPLANTAR KERATODERMAS AND RELATED CONDITIONS
PALMOPLANTAR KERATODERMAS
Unna–Thost syndrome
Greither’s syndrome
Olmsted’s syndrome
Vohwinkel’s syndrome
Epidermolytic palmoplantar keratoderma (Vörner’s syndrome)
Howel–Evans syndrome
Papillon–Lefèvre syndrome
Haim–Munk syndrome
Mal de Meleda
Mal de Naxos (Naxos disease)
Carvajal syndrome
Gamborg Nielsen keratoderma
Palmoplantar keratoderma with sclerodactyly (Huriez syndrome)
Punctate palmoplantar keratoderma
Striate palmoplantar keratoderma
Circumscribed keratoderma
Acquired keratoderma
Aquagenic acrokeratoderma
Histopathology
Interdigital keratoderma
Histopathology of palmoplantar keratodermas
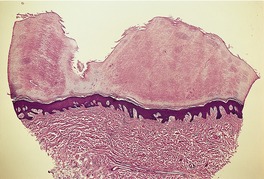
Fig. 9.6
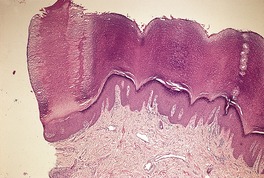
Fig. 9.7
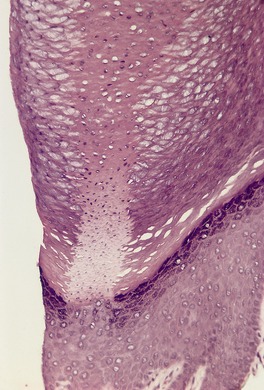
Fig. 9.8
Electron microscopy
OCULOCUTANEOUS TYROSINOSIS
Histopathology658
ACROKERATOELASTOIDOSIS
Histopathology685
Electron microscopy
PACHYONYCHIA CONGENITA
Histopathology
CORNOID LAMELLATION
POROKERATOSIS AND VARIANTS
Porokeratosis of Mibelli (including site-specific variants of face, scalp, and anogenital region)
Linear and systematized porokeratosis
Hyperkeratotic verrucous porokeratosis
Disseminated superficial actinic porokeratosis (types 1, 2, and 3)
Disseminated eruptive porokeratosis
Follicular porokeratosis
Inflammatory porokeratosis
Eruptive pruritic porokeratosis
Palmoplantar porokeratoses:
Porokeratosis plantaris discreta
Porokeratotic palmoplantar keratoderma discreta
Punctate porokeratotic keratoderma (punctate porokeratosis)
Porokeratosis punctata palmaris et plantaris (spiny keratoderma)
Linear palmoplantar porokeratosis
Porokeratosis plantaris, palmaris et disseminate
Porokeratotic eccrine ostial and dermal duct nevus
Porokeratotic eccrine duct and hair follicle nevus
Porokeratoma
CAP syndrome (craniosynostosis, anal anomalies)
Treatment of porokeratosis
Histopathology
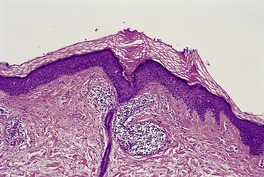
Fig. 9.9
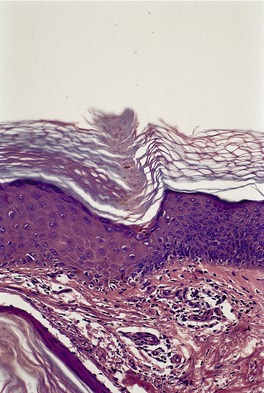
Fig. 9.10
Electron microscopy
EPIDERMOLYTIC HYPERKERATOSIS
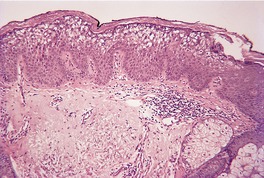
Fig. 9.11
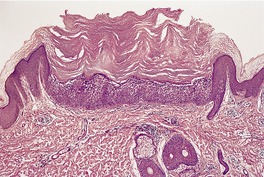
Fig. 9.12
Electron microscopy
EPIDERMOLYTIC ACANTHOMA
Histopathology
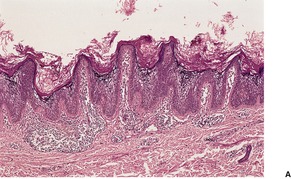
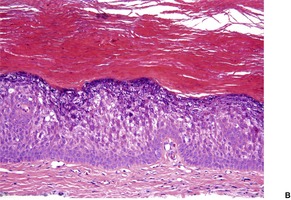
Fig. 9.13
ACANTHOLYTIC DYSKERATOSIS
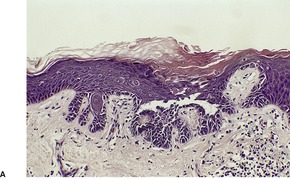
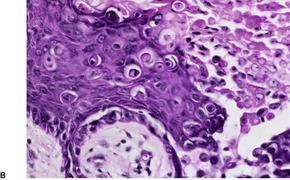
Fig. 9.14
FOCAL ACANTHOLYTIC DYSKERATOSIS
Histopathology
Acantholytic subset
Dyskeratotic subset
DARIER’S DISEASE
Treatment of Darier’s disease
Histopathology
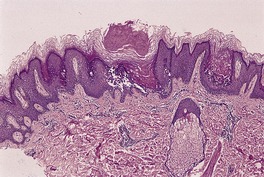
Fig. 9.15
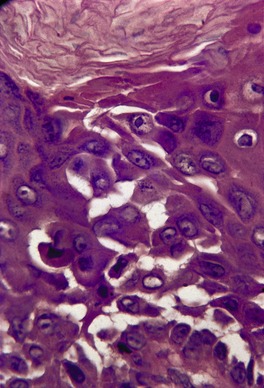
Fig. 9.16
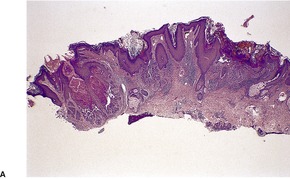
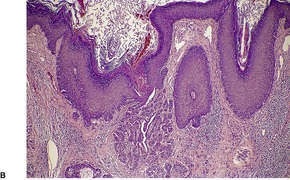
Fig. 9.17
Electron microscopy
GALLI–GALLI DISEASE
Histopathology
GROVER’S DISEASE
Treatment of Grover’s disease
Histopathology
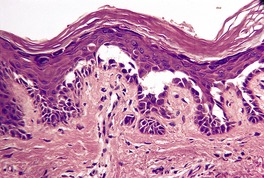
Fig. 9.18
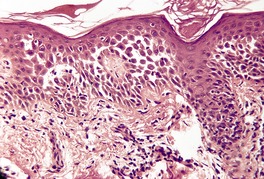
Fig. 9.19
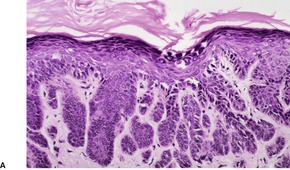
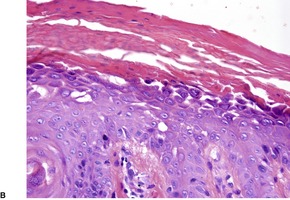
Fig. 9.20
Electron microscopy
HAILEY–HAILEY DISEASE
Treatment of Hailey–Hailey disease
Histopathology
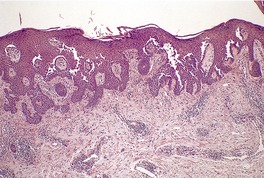
Fig. 9.21
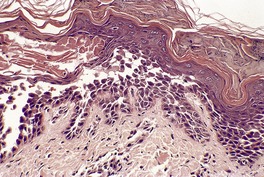
Fig. 9.22
Electron microscopy
WARTY DYSKERATOMA
Histopathology1228. and 1230.
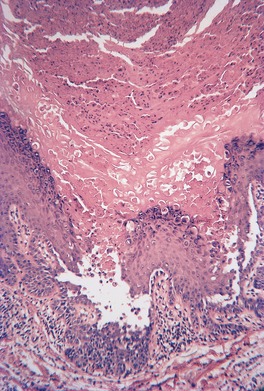
Fig. 9.23
HYPERGRANULOTIC DYSCORNIFICATION
Histopathology1237
COLLOID KERATOSIS
Histopathology1239
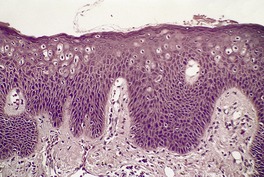
Fig. 9.24
DISCRETE KERATOTIC LESIONS
HYPERKERATOSIS LENTICULARIS PERSTANS
Histopathology1246.1251. and 1256.
Electron microscopy
KYRLE’S DISEASE
Histopathology1283. and 1284.
MULTIPLE MINUTE DIGITATE KERATOSES
Histopathology
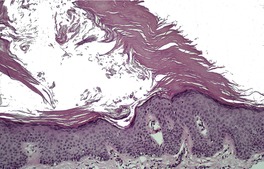
Fig. 9.25
WAXY KERATOSES
Histopathology
MISCELLANEOUS EPIDERMAL GENODERMATOSES
ACROKERATOSIS VERRUCIFORMIS
Histopathology1315. and 1318.
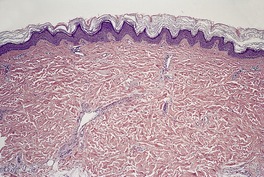
Fig. 9.26
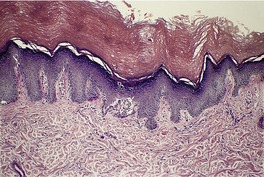
Fig. 9.27
XERODERMA PIGMENTOSUM
Type
OMIM
Gene defect
Gene locus
XP-A
278700
XPA
9q22.3
XP-B
133510
XPB (ERCC3)
2q21
XP-C
278720
XPC
3p25
XP-D
278730
XPD (ERCC2)
19q13.2–q13.3
XP-E
278740
XPE (DDB2)
11p12–p11
XP-F
278760
XPF (ERCC4)
16p13.3–p13.13
XP-G
278780
XPG (ERCC5)
13q33
XP variant
278750
POLH
6p21.1–p12
De Sanctis–Cacchione syndrome
278800
XPA/ERCC6
9q22.3/10q11
Histopathology1327
Electron microscopy
ECTODERMAL DYSPLASIAS
Anhidrotic (hypohidrotic) ectodermal dysplasia
Ectodermal dysplasia/skin fragility syndrome
Hidrotic ectodermal dysplasia
Orofaciodigital syndrome
Cardiofaciocutaneous (CFC) syndrome
Ectodermal dysplasias with clefting
Histopathology of the ectodermal dysplasias
CUTANEOUS AND MUCOSAL DYSKERATOSIS
NEVOID HYPERKERATOSIS OF THE NIPPLE
Histopathology
PEELING SKIN SYNDROME
Histopathology
MISCELLANEOUS DISORDERS
GRANULAR PARAKERATOSIS
Histopathology
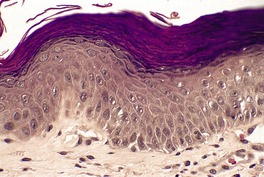
Fig. 9.28
CIRCUMSCRIBED ACRAL HYPOKERATOSIS
Histopathology
Electron microscopy
WHITE SPONGE NEVUS
Histopathology
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Disorders of epidermal maturation and keratinization
Ichthyoses249
Ichthyosis vulgaris249
X-linked ichthyosis250
Bullous ichthyosis252
Netherton’s syndrome253
Erythrokeratodermia variabilis253
Harlequin ichthyosis254
Follicular ichthyosis255
Acquired ichthyosis255
Refsum’s disease255
Other ichthyosis-related syndromes255
Sjögren–Larsson syndrome255
‘KID’ syndrome255
Conradi–Hünermann–Happle syndrome256
Neu–Laxova syndrome256
‘CHILD’ syndrome256
‘IBIDS’ (ichthyosis with trichothiodystrophy)256
‘ILVASC’256
Multiple sulfatase deficiency256
‘MAUIE’ syndrome256
Neutral lipid storage disease (Dorfman–Chanarin syndrome)256
Shwachman syndrome257
Palmoplantar keratodermas and related conditions257
Palmoplantar keratodermas257
Unna–Thost syndrome257
Greither’s syndrome257
Olmsted’s syndrome257
Vohwinkel’s syndrome258
Epidermolytic palmoplantar keratoderma (Vörner’s syndrome)258
Howel–Evans syndrome258
Papillon–Lefèvre syndrome258
Haim–Munk syndrome258
Mal de Meleda258
Mal de Naxos (Naxos disease)258
Carvajal syndrome259
Gamborg Nielsen keratoderma259
Palmoplantar keratoderma with sclerodactyly (Huriez syndrome)259
Punctate palmoplantar keratoderma259
Striate palmoplantar keratoderma259
Circumscribed keratoderma259
Acquired keratoderma259
Aquagenic acrokeratoderma260
Interdigital keratoderma260
Oculocutaneous tyrosinosis260
Acrokeratoelastoidosis261
Pachyonychia congenita261
Hypergranulotic dyscornification272
Colloid keratosis272
This chapter deals with a heterogeneous group of diseases in which an abnormality of maturation, of keratinization, or of structural integrity of the epidermis is present. Many advances have been made in the last decade in our understanding of the molecular basis of these disorders. Most of these conditions are genetically determined, although a few are acquired diseases of adult life. An understanding of these disorders requires a knowledge of the structure of the epidermis and the process of normal keratinization.
The epidermis is a stratified squamous epithelial sheet covering the external surface of the body. It is composed of keratinocytes and melanocytes forming a binary system. 1 The keratinocytes are continuously regenerating with the cells undergoing terminal differentiation and death. Folds of the epidermis (the rete ridges) extend into the dermis while the dermis projects upwards between these ridges, the so-called ‘dermal papillae’. The epidermis is separated from the dermis by the basement membrane, a complex, multilayered structure that contributes to the structural framework of the epidermis (see p. 140).
Resting on the basement membrane is the basal layer of the epidermis, which contains the proliferating cells of the epidermis. Normally, only about 17% of the basal cells make up the dividing cell population. Cells leave this layer to undergo terminal differentiation, whereas some immediately die by apoptosis as a result of an intrinsic program or imbalance of signaling factors. 2 Cells destined to differentiate enter the prickle cell layer, where they acquire more cytoplasm and well-formed bundles of keratin intermediate filaments (tonofilaments). The major function of the keratin filaments is to endow epithelial cells with the mechanical resilience they need to withstand stress. 3 The intercellular attachments, the prickles or desmosomes, develop here. There is also a change in the keratin composition of the keratin intermediate filaments; the keratins K1 and K10 are expressed in suprabasal cells, in contrast to K5 and K14 in the basal layer (see below). The prickle cell layer varies in thickness from 4 to 10 cells. As the cells are pushed outwards they begin synthesizing the proteins that eventually constitute the keratohyaline granules and cell envelope of the granular layer and stratum corneum respectively. The granular layer or stratum granulosum is identified by the presence of the keratohyaline granules. This layer is 1–3 cells thick. The cells lose their cytoplasmic organelles and metabolic activity. They flatten further and become compacted into a dense keratinous layer known as the stratum corneum. The superficial flake-like squames are eventually cast off (desquamate).
The total epidermal renewal time is approximately 2 months. The cells take 26–42 days to transit from the basal layer to the granular layer and a further 14 days to pass through the keratin layer.
Keratinization refers to the cytoplasmic events that occur in the cytoplasm of epidermal keratinocytes during their terminal differentiation. It involves the formation of keratin polypeptides and their polymerization into keratin intermediate filaments (tonofilaments). It is estimated that each keratin intermediate filament contains 20 000–30 000 keratin polypeptides. More than 30 different keratins have been identified – more than 20 epithelial keratins and 10 hair keratins.4. and 5. The epithelial keratins are divided by molecular weight and isoelectric points into two types – type I keratins, which are acidic and of lower molecular weight, and type II keratins, which are neutral basic. The type I keratins are further subdivided numerically from K10 to K20 and the type II keratins from K1 to K9. As a general rule, the epithelial keratins are coexpressed in specific pairings with one from each type. 4 For example, in the basal layer the keratins are K5 and K14 and in the suprabasal layers K1 and K10. K15 has no defined type II partner. 6 Keratins additional to a pair are sometimes found. For example, K6, K16, and K17 are found in the nail bed epithelium. A new keratin, designated K6irs, has been localized to the inner root sheath of the hair follicle. 7
Keratins exhibit a high degree of tissue specificity. The primary location of each of the keratin types is shown in Table 9.1, which is based on Irvine and McLean’s work, 8 and a later paper by Chu and Weiss. 9 Keratin mutations can cause epithelial fragility syndromes, such as epidermolysis bullosa simplex, in addition to some of the ichthyoses and keratodermas. 8 The various disorders of keratin and their corresponding keratin mutation are listed in Table 9.2.
The keratin intermediate filaments aggregate into bundles (tonofilaments) which touch the nuclear membrane and extend through the cytoplasm to interconnect with adjacent cells, indirectly, via the desmosomal plaques.4. and 10.
The keratohyaline granules which give the identifying features to the granular layer result from the accumulation of newly synthesized proteins. One of these is profilaggrin. It undergoes dephosphorylation to form filaggrin, a histidine-rich protein which acts as a matrix glue and facilitates filament aggregation into even larger bundles. Filaggrin rapidly aggregates the keratin cytoskeleton, causing collapse of the granular cells into flattened anuclear squames. 11 Trichohyalin, found primarily in the inner root sheath cells, is sometimes coexpressed with filaggrin. 12 The keratin filaments are stabilized by disulfide crosslinks which make this intracytoplasmic structural mesh highly insoluble. A second polypeptide, loricrin, is localized to the keratohyaline granules. It contributes to the formation of a stable, intracytoplasmic, insoluble barrier known as the cell envelope (cornified envelope) – see below.
The granular layer also contains small, lipid-rich lamellated granules (100–500 nm in diameter), known as Odland bodies or membrane-coating granules. They are secreted into the intercellular space in this region. Their lipid-rich nature contributes to the permeability barrier (see below). 13
The cell envelope (cornified envelope) forms just beneath the cell membrane. It is 7–15 nm wide and composed of crosslinked proteins. Several proteins are involved in the formation of the cell envelope including loricrin, involucrin, keratolinin, and small proline-rich proteins.4. and 14. Polymerization and crosslinking of these proteins requires the action of calcium-dependent epidermal transglutaminases, of which three have been identified in the skin. The heat shock protein hsp27 appears to play a role in the assembly of the cornified cell envelope. 15
Keratinocytes in the stratum corneum (corneocytes) are dead. They eventually undergo desquamation, an orderly process in which individual corneocytes detach from their neighbors at the skin surface and are swept away. 16 This occurs, in part, because the desmosomes are degraded (presumably by proteases) during transit through the stratum corneum. 17 One of these enzymes is stratum corneum chymotryptic enzyme, a serine protease which causes proteolysis of desmosomes in the stratum corneum. It is reduced, but not absent, in the ichthyoses. 18 However, the process of desquamation (dyshesion) is more complex than simple desmosomal degeneration. 19 It is known that cholesterol esters are important components of cell adhesion. For example, in X-linked ichthyosis, there is an accumulation of cholesterol sulfate associated with a deficiency in aryl sulfatase, which results in decreased desquamation and keratin accumulation. It is thought that cholesterol sulfate inhibits proteases that are involved in desquamation. 20 Lipids also play a role in the permeability barrier of the skin, a function which resides in the region of the granular layer.21. and 22. Lipids secreted by the Odland bodies (see above) are an important component of the permeability barrier. In addition to the structures already described, there is a skin surface lipid film produced by secreted sebum mixed with lipid from the keratinizing epithelium. 23 It contributes to barrier function.
The chromosomal localizations of the various genes encoding the various polypeptides involved in keratinization have been elucidated – type I keratins on chromosome 17, type II on chromosome 12, transglutaminases on chromosome 14, and profilaggrin, trichohyalin, loricrin, involucrin, and the small proline-rich proteins on chromosome 1q21. 4 Because this latter gene complex controls the structural proteins of cornification, the term ‘epidermal differentiation complex’ has been proposed for this region. 14 The expression of these genes is controlled by proteins called transcription factors.24. and 25.
The abnormal process, where known, for each of the disorders of keratinization is shown in Table 9.3. This is based on a publication by Hohl. 26
Cell envelope
(transglutaminase-1)
(transglutaminase-5)
The ichthyoses are a heterogeneous group of hereditary and acquired disorders of keratinization characterized by the presence of visible scales on the skin surface.27.28.29. and 30. The word is derived from the Greek root for fish – ichthys. There are four major types of ichthyosis: ichthyosis vulgaris, X-linked ichthyosis, ichthyosis congenita, and epidermolytic hyperkeratosis (bullous ichthyosiform erythroderma). In addition, there are several rare syndromes in which ichthyosis is a major feature. It has been estimated that nearly 1 million Americans have either ichthyosis vulgaris or X-linked recessive ichthyosis, the most common forms. 31 For patients, family, and friends wanting information this can be found on the website of The Foundation for Ichthyosis and Related Skin Types at www.scalyskin.org. 32
Kinetic studies have shown that lamellar ichthyosis and epidermolytic hyperkeratosis are characterized by hyperproliferation of the epidermis with transit times of 4–5 days, whereas the scale in ichthyosis vulgaris and X-linked ichthyosis is related to prolonged retention of the stratum corneum (retention hyperkeratoses).17.27. and 28. This may be related to a persistence of desmosomes in the stratum corneum. 33
Although the mode of inheritance was originally used as a major criterion in the delineation of the various forms of ichthyosis, recent studies have shown some evidence of genetic heterogeneity in the various groups.34. and 35. Consanguinity is an important association in some communities.36. and 37. Our understanding of the molecular basis of many of the ichthyoses has progressed tremendously in the last decade. 38
Oral retinoid therapy is often used in the treatment of disorders of keratinization including the ichthyoses. A retrospective study involving patients on this treatment for up to 25 years reported disappointing results with nearly 50% of patients experiencing no benefit ± side effects. 39 Work is ongoing to develop a drug for the ichthyoses with a more favorable tolerability profile than retinoids such as acitretin. One such drug may be liarozole, an imidazole given orphan drug status by the European Commission and the FDA in the US. 40 In a phase II/III, double-blind, randomized trial liarozole is equally effective as a treatment for ichthyosis as acitretin, but shows a trend towards a more favorable tolerability profile. 40
Ichthyosis vulgaris (OMIM 146700) is the most common disorder of keratinization (incidence 1 : 250), with an onset in early childhood and an autosomal dominant inheritance.27. and 37. Heterozygotes show a very mild phenotype with incomplete penetrance (semidominant); such cases may be misdiagnosed as dry skin.11. and 41. The disorder is lifelong. It is characterized by fine, whitish scales involving particularly the extensor surfaces of the arms and legs, as well as the scalp. Flexures are spared. There may be accentuation of palmar and plantar markings, keratosis pilaris, and features of atopy.
In ichthyosis vulgaris, there is a deficiency in profilaggrin which is converted into filaggrin, the major protein of keratohyalin.17. and 42. There are quantitative decreases in filaggrin, related to the severity of the disease. 43 This results from loss-of-function mutations in the gene encoding filaggrin (FLG), located at 10q21.11.44.45. and 46. The gene is unusually large and repetitive, making analysis of it difficult. 11 Mutations in the FLG gene occur in approximately 9% of individuals from European populations. 47 Childhood eczema is strongly associated with these common European mutations.48. and 49. A previously unreported genetic locus for ichthyosis vulgaris has been identified in two Chinese families on chromosome 10q22.3–q24.2. 50 No specific gene has, as yet, been identified.
The treatment of ichthyosis vulgaris will depend on the severity of the disease. In mild cases emollients and keratolytics may suffice. Hydration can be achieved with 12% ammonium lactate cream or lotion and keratolysis with a 6% salicylic acid gel. Many products on the market contain urea or propylene glycol and benefit these patients. 51 Topical retinoids may also be beneficial. Future therapy may be directed towards compounds that increase filaggrin expression in keratinocytes, such as oleanolic acid and ursolic acid, and other products in medicinal herbs and plants. 47
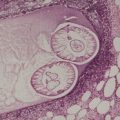
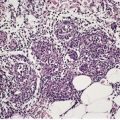
The epidermis may be of normal thickness or slightly thinned with some loss of the rete ridges. 52 There is mild to moderate hyperkeratosis, associated paradoxically with diminution or absence of the granular layer (Fig. 9.1). The thickened stratum corneum is often laminated in appearance. The hyperkeratosis may extend into the hair follicles. The sebaceous and sweat glands are often reduced in size and number.
Ichthyosis vulgaris. There is a thickened layer of compact orthokeratosis overlying a diminished granular layer. (H & E)
Electron microscopy shows defective keratohyaline synthesis with small granules having a crumbled or spongy appearance. 42
This form of ichthyosis (OMIM 308100), which is inherited as an X-linked recessive trait, is present at birth or develops in the first few months of life. It is characterized by large polygonal scales which are dirty brown in color and adherent. X-linked ichthyosis may involve the entire body in varying degree, although there is sparing of the palms and soles. The preauricular region is characteristically involved in this variant of ichthyosis; in contrast, this site is usually spared in ichthyosis vulgaris. 53 Corneal opacities and mental retardation may also occur in X-linked ichthyosis.54.55.56. and 57. Other rare associations include congenital dislocation of the hip, 58 Poland’s syndrome (unilateral absence of the pectoralis major muscle – OMIM 173800) 59 and Kallmann’s syndrome (hypogonadism, renal agenesis, anosmia and synkinesis – OMIM 308700). 60 Interestingly, X-linked ichthyosis and Kallmann’s syndrome result from abnormalities in contiguous genes. 60 It has also been associated with the oculo-auriculo-vertebral spectrum (Goldenhar syndrome – OMIM 164210). 61 Ichthyosis vulgaris and X-linked ichthyosis have been reported in the same family. 62 Male-pattern baldness does occur, despite earlier claims to the contrary.63. and 64.
Its incidence is approximately one in 5000–6000 males, but occasionally female heterozygotes have mild scaling of the legs. 57 Full expression of the disease has only been reported in a few females. 65
This condition is characterized by a deficiency of steroid sulfatase in a wide range of tissues, including leukocytes and fibroblasts. As a result there is an accumulation of cholesterol sulfate in the pathological scales and in serum and leukocytes.16.66.67.68. and 69. Accumulation in the skin interferes with normal barrier function70. and 71. and with proteases involved in the dissolution of desmosomes required for desquamation of the stratum corneum. 20 There are elevated levels of cholesterol sulfate and dehydroepiandrosterone in the serum. 72 The deficiency in placental sulfatase which is also present results in decreased maternal urinary estrogens and a failure to initiate labor in some cases. 54 The steroid sulfatase (STS) gene is on the distal short arm of the X chromosome (Xp22.3). 55 Most cases involve complete or partial deletion of the STS gene and flanking sequences.73.74.75.76.77.78.79. and 80. The defect is usually inherited and it is not due to a de novo mutation. 81 In most cases, there appears to be paternal transmission of the affected X chromosome. 80 FISH analysis is best performed for an accurate diagnosis of the female carrier state. 82
Treatment regimens similar to those for ichthyosis vulgaris can be used.
There is usually conspicuous acanthosis with thickening of the stratum corneum and a normal to thickened granular layer. Acanthosis was absent in all nine cases studied in one series. 83 Thinning of the granular layer is sometimes present. 84 There is often hyperkeratosis of follicular and sweat duct orifices. 85 Suprabasal and basal vacuolation are sometimes present. 83 There may be a few lymphocytes around the vessels in the superficial dermis.
In recent years there has been an attempt to reclassify the autosomal recessive ichthyoses previously known as lamellar ichthyosis (LI) and non-bullous congenital ichthyosiform erythroderma (CIE) as ichthyosis congenita on the basis of ultrastructural findings suggesting that there are at least four types.86.87. and 88. It has an estimated prevalence of one in 300 000 newborns and most of the infants are born as ‘collodion babies’. 89 This new classification has not yet been widely accepted by clinicians; furthermore, inheritance and ultrastructure have not always proven to be a reliable basis for classifications. A defect in keratinocyte transglutaminase has been detected in about 50% of the families studied. 90
To date, six genes have been identified for autosomal recessive congenital ichthyosis. They include TGM1 on chromosome 14q11, ABCA12 on chromosome 2q34–q35, 91ichthyin on chromosome 5q33, ALOXE3 and ALOX12B on chromosome 7p13, and CYP4F22 (FLJ39501) on chromosome 19p12–q12.89. and 92. Two additional loci have been identified, the first on chromosome 19p13.2–p13.1 (? CYP4F22) and the second on chromosome 12p11.2–q13. 89 In addition, mutations in CG1-58/ABHD5 on chromosome 3p21 have been found to underlie Chanarin–Dorfman syndrome (OMIM 275630). 89 Another syndromic congenital ichthyosis is hypotrichosis, caused by a mutation in ST14 on 11q24.3–q25. The gene encodes the serum protease matriptase. 93 The genes in the LOX cluster are involved in the subset of congenital ichthyosis known as (non-bullous) congenital ichthyosiform erythroderma. 89
The four subtypes of ichthyosis congenita are discussed below. This classification may need to be modified or abandoned as overlapping ultrastructural features have been reported between some of the types. 94
Type I is the largest group and corresponds to congenital ichthyosiform erythroderma (CIE) of the older classification (see below). There is erythroderma with fine scaling. About 40% present as a ‘collodion baby’, in which the infant is encased in a tight membrane. 88 Most have palmoplantar keratoderma. Clear ultrastructural criteria are lacking but numerous lipid droplets are usually present in the horny cells. 87 It has been suggested that the diagnosis be made by exclusion of the other three types. This group may still be heterogeneous.
Type II is characterized by cholesterol clefts in the horny cells. It corresponds to lamellar ichthyosis (see below).
Lamellar ichthyosis (OMIM 242300) is characterized by large plate-like scales of ichthyosis. 98 It corresponds to ichthyosis congenita type II (see above). It may present as a ‘collodion baby’.28.99.100.101. and 102. The palms and soles are often involved. 28 Nail plates are also affected. 103 Pseudoainhum has also been reported. 104 It accounted for 5.6% of all ichthyoses in one series from Saudi Arabia. 36 An increased incidence of skin cancers has been reported in this variant of ichthyosis. 105 It has also been associated with rickets, 106 25-hydroxyvitamin D deficiency, 107 and with dermatophytosis. 108
This variant has been shown to have mutations in the keratinocyte transglutaminase-1 (TGM1) gene on chromosome 14q11.1.109.110.111.112.113.114.115.116. and 117. Plasminogen activator inhibitor-2, a substrate of transglutaminase-1, has normal expression in a large group of TGase-1-proficient congenital ichthyosis, making it unlikely that this inhibitor is a primary molecular cause of this type of ichthyosis. 118 This defect in transglutaminase-1 interferes with the crosslinkage of loricrin and involucrin and the formation of the cornified cell envelope. 119
A second variant of lamellar ichthyosis (OMIM 601277) is due to mutations in the ABCA12 gene (an ATP binding cassette-4-protein) which has been mapped to chromosome 2q34–q35.
Congenital ichthyosiform erythroderma (CIE) was known in the past as non-bullous congenital ichthyosiform erythroderma (OMIM 242100). It has also been included within the category of lamellar ichthyosis. CIE is known to be a heterogeneous entity; 120 variants have been reclassified as ichthyosis congenita types I, III, and IV (see above). Consanguinity is high. 121 Variants include a pustular form122 and a reticulated pigmented form (ichthyosis variegata).123. and 124. Most present as collodion babies. 121 It should be mentioned that loricrin keratoderma (see p. 254) can also present as a collodion baby and mimic congenital ichthyosiform erythroderma. 125 A patient with associated ocular albinism and Noonan’s syndrome has been reported. 126 Multiple aggressive squamous cell carcinomas developed in one patient with CIE. 127 CIE differs from lamellar ichthyosis (LI) by the erythroderma and finer, pale scales which have a high content of n-alkanes.67.128. and 129. A subset of patients with CIE may have abnormal expression of keratinocyte transglutaminase-1, 130 but the genes most commonly implicated in this variant are part of the LOX gene cluster, ALOXE3 and ALOX12B on chromosome 17p1389.131.132. and 133. and ABCA12 on 2q34–q35. 91
Oral retinoids have been used to treat this condition. 134
Some of the reports in the literature have not distinguished between lamellar ichthyosis and congenital ichthyosiform erythroderma or the various subtypes of ichthyosis congenita; a composite description therefore follows. However, in the study from the Great Ormond Street Hospital in London, non-bullous ichthyosiform erythroderma and lamellar ichthyosis were histologically indistinguishable. 83
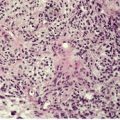
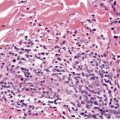
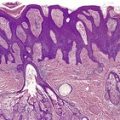
There is hyperkeratosis, focal parakeratosis, and a normal or thickened granular layer. 135 The hyperkeratosis is more marked in lamellar ichthyosis than other variants, and there is easily discernible parakeratosis in CIE. 128 There is often some acanthosis and occasionally there is irregular psoriasiform epidermal hyperplasia (Fig. 9.2 and Fig. 9.3). 35 Vacuolation of cells in the granular layer is a rare finding. 136 Keratotic plugging of follicular orifices may also be present. The dermis often shows a mild superficial perivascular infiltrate of lymphocytes.
Lamellar ichthyosis. There is compact orthokeratosis and mild psoriasiform hyperplasia of the epidermis. (H & E)
Lamellar ichthyosis. Another case with more orthokeratosis and a less regular outline. (H & E)
The case presented recently with ichthyosiform erythroderma, onset in adolescence, and a lichenoid tissue reaction on histology probably represents a new entity. 137
The ultrastructural features of the various types of ichthyosis congenita (including LI and CIE) have been outlined above.
The term ‘bullous ichthyosis’ is preferred to bullous congenital ichthyosiform erythroderma (OMIM 113800), and to epidermolytic hyperkeratosis (which merely describes a histological reaction pattern).138.139. and 140. Despite this confusion in terminology there is a resurgence in the use of the term epidermolytic hyperkeratosis.141. and 142. Bullous ichthyosis is a rare, autosomal dominant condition which is usually severe and characterized at birth by widespread erythema and some blistering. 28 Coarse, verrucous scales, particularly in the flexures, develop as the disposition to blistering subsides during childhood. Annular plaques, of late onset, have been reported. 143 It has been associated with hypocalcemic vitamin D-resistant rickets. 144
Bullous ichthyosis is a clinically heterogeneous condition.145.146.147.148.149. and 150. Six subtypes were recently defined, the presence or absence of palmoplantar keratoderma being used as a major feature in defining the subtypes. 151 There is some evidence to suggest that cases with severe palmoplantar keratoderma (type PS-1) have abnormalities in KRT1, while those without have abnormalities in KRT10.142.152.153. and 154. In cases of palmoplantar keratoderma without diffuse cutaneous lesions, defects in KRT9 are usual. The commonest abnormality in KRT10 involves an arginine to histidine substitution at one point.155. and 156. Other substitutions have been reported.157.158. and 159. No abnormality in KRT1 or KRT10 has been detected in some cases, suggesting that other genes may be involved. Keratin 1 and 10 are coexpressed to form keratin intermediate filaments in the suprabasal layers of the epidermis.149.151.160.161.162. and 163. The KRT1 and KRT10 genes are located on chromosome 12 in the type II keratin cluster. Approximately 50% of cases involve spontaneous mutations in either gene. 164 Clinical variants of bullous ichthyosis include a rare acral group (not included in the six subtypes mentioned above), 10 an annular form (due in one case to a KRT10 mutation),161.165. and 166.ichthyosis hystrix of Curth–Macklin (OMIM 146590) due to mutations in the keratin 1 (KRT1) gene at 12q13, and characterized ultrastructurally by perinuclear shells of unbroken tonofilaments), 167ichthyosis hystrix of Lambert,168. and 169. and ichthyosis bullosa of Siemens.170.171.172.173.174.175.176.177.178. and 179. Ichthyosis bullosa of Siemens (OMIM 146800) is due to mutations in the keratin 2e gene (KRT2) at 12q11–q13, in contrast to the mutations in keratin 1 or 10 in patients with epidermolytic hyperkeratosis/bullous congenital ichthyosiform erythroderma.180. and 181. It is autosomal dominant. The hyperkeratosis is usually limited to the flexural areas and there is no erythroderma. 182 There is circumscribed shedding of the stratum corneum over hyperkeratotic skin (the Mauserung phenomenon).182.183. and 184.Ichthyosis exfoliativa (exfoliative ichthyosis – OMIM 607936) is said to have a similar genetic defect and clinical features to ichthyosis bullosa of Siemens, but one report has suggested that there is no epidermolytic hyperkeratosis on histology.185. and 186. It also appears to be autosomal recessive with linkage to 12q13.187. and 188.
Prenatal diagnosis can be made at approximately 19 weeks by fetal skin biopsy examined by light and electron microscopy; 189 it can be made earlier (10–11 weeks’ gestation) by direct gene sequencing of chorionic villus samples.190. and 191.
Retinoid therapy is particularly effective in patients with KRT10 mutations, but not in patients with KRT1 mutations. 192 The mosaic form of this disease has been successfully treated with topical maxacalcitol, a vitamin D3 analogue with approximately 10 times greater efficacy at suppressing keratinocyte proliferation in vitro than calcipotriol. 193
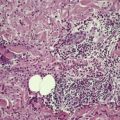
The histological pattern is that of epidermolytic hyperkeratosis, characterized by marked hyperkeratosis and granular and vacuolar change in the upper spinous and granular layers (Fig. 9.4). 194 Intracytoplasmic and perinuclear eosinophilic homogenizations are identified with varying frequencies. They correspond to the aggregation of tonofilaments seen on electron microscopy. 195 The keratohyaline granules appear coarse and basophilic with clumping. There is moderate acanthosis. The histological features of blistering can sometimes be subtle, with only slight separation of the markedly vacuolar cells in the mid and upper epidermis. There is usually a mild perivascular inflammatory cell infiltrate in the upper dermis.
Bullous ichthyosis with hyperkeratosis and granular and vacuolar change of the keratinocytes in the upper layers of the epidermis. Blistering is not shown in this field. (H & E)
In ichthyosis bullosa of Siemens the changes are usually confined to the granular layer and the superficial spinous cells. 182
There is aggregation of tonofilaments at the cell periphery with perinuclear areas free of tonofilaments and containing endoplasmic reticulum.196. and 197. In the upper granular layer there are numerous keratohyaline granules, sometimes embedded in clumped tonofilaments. Although desmosomes appear normal, there is often an abnormality in the association of tonofilaments and desmosomes.
Netherton’s syndrome (OMIM 256500) is a rare autosomal recessive disease characterized by the triad of ichthyosis, trichorrhexis invaginata (bamboo hair), and an atopic diathesis with elevated levels of IgE.198.199.200.201.202.203.204.205. and 206. Infants are usually born with ichthyosiform erythroderma which may persist, 207 or eventually become a milder, migratory and polycyclic ichthyosis known as ichthyosis linearis circumflexa (ILC). Patches of ILC do not usually appear until after the first year of life, sometimes delaying the diagnosis.208.209.210. and 211. Other clinical manifestations include intermittent aminoaciduria, elevated serum levels of interleukin 18 (IL-18), mental retardation, enteropathy, and hemihypertrophy.212. and 213.
Netherton’s syndrome is due to mutations of SPINK5 on chromosome 5q32.214. and 215. It is telomeric to the cytokine gene cluster at 5q31. 216SPINK5 encodes the lymphoepithelial-Kazal-type-related inhibitor (LEKTI), a serine protease inhibitor which has a crucial role in epidermal growth and differentiation.209.214.215.217. and 218. An immunohistochemical test for the presence of LEKTI in skin has been developed. It is absent in Netherton’s syndrome. 219 There is some correlation between the mutations and the phenotype. 220
Other hair shaft abnormalities such as pili torti, trichorrhexis nodosa, and monilethrix have also been reported in Netherton’s syndrome.221. and 222. The hair shaft abnormalities are more common in eyebrow than scalp hair. 223 Hair shaft changes may not appear until 18 months of age. 224 The diagnosis has been made by dermoscopy of the hairs in one suspected case; trichorrhexis invaginata was present. 225
Disturbances in the immune system have been reported in several patients with Netherton’s syndrome. 226 This may be the explanation for the finding of infections, including HPV infection, superimposed on the ichthyotic lesions.227. and 228. Squamous cell carcinomas may develop. 229 CD30+ T-cell lymphoma developed in one patient with Netherton’s syndrome who underwent a heart transplant for idiopathic cardiomyopathy. 217 Cases with phenotypic overlap with the peeling skin syndrome (see p. 278) have been reported.230. and 231. The keratin filaments from the scales in Netherton’s syndrome are composed of reduced amounts of high molecular subunits and increased amounts of low molecular subunits. 198 Some patients may have ichthyosis linearis circumflexa in the absence of features of Netherton’s syndrome.
Various treatments have been used with variable success. Emollients are useful adjuncts with other therapies. Topical corticosteroids are moderately effective, but their long-term use is contraindicated. Topical retinoids may eventually result in aggravation of the condition. Topical calcipotriol, PUVA, cyclosporine (ciclosporin), and ammonium lactate 12% lotion have also been used. 232 Good control was obtained with tacrolimus and pimecrolimus in one report, 232 but another found no benefit from tacrolimus. 206
The skin lesions show hyperkeratosis, a well-developed granular layer, and acanthosis. The margin shows focal parakeratosis with absence of the granular layer and more obvious psoriasiform epidermal hyperplasia (Fig. 9.5).198.233. and 234. In erythrodermic cases, the parakeratosis is more prominent; sometimes it constitutes the entire stratum corneum. In all cases, the outermost nucleated layers do not flatten normally. 207 Pustules are sometimes seen in the erythrodermic stage. Epidermal mitoses are increased. In some cases PAS-positive, diastase-resistant granules can be found in the prickle cells. There is often a mild perivascular inflammatory cell infiltrate in the superficial dermis.
Ichthyosis linearis circumflexa. This biopsy from a neonate shows a striking resemblance to psoriasis, but there are no mounds of parakeratosis containing neutrophils. (H & E)
Erythrokeratodermia variabilis (EKV – OMIM 133200) is a rare, usually autosomal dominant form of ichthyosis that develops in infancy; uncommonly it is present at birth. 235 A rare autosomal recessive variant is now recognized. 236 There are transient erythematous patches and erythematous hyperkeratotic plaques which are often polycyclic or circinate.27.32.237. and 238. Lesions may resemble erythema gyratum repens. 239 A targetoid appearance is seen in the rare cocarde variant.240. and 241. Follicular hyperkeratosis has been described. 242 There is retention hyperkeratosis associated with a basal cell type of keratin. 243
The disorder has been mapped to chromosome 1p35.1, but is genetically heterogeneous. EKV may be caused by mutations in one of two neighboring connexin genes, GJB3 and GJB4, encoding the gap junction proteins Cx31 and Cx30.3 respectively.243.244.245.246.247. and 248. Sporadic cases also occur. Cases with no identifiable gene defect have been reported. 249 Cx31 is expressed predominantly in the stratum granulosum in normal skin. 250 The ‘new’ type of erythrokeratoderma reported by van Steensel et al251 has subsequently been reclassified as KLICK syndrome (OMIM 601952).252. and 253.
Progressive symmetric erythrokeratodermia (OMIM 602036), thought at one time to be the same condition, appears to be a variant with its own specific abnormality, a mutant loricrin gene.26. and 254. Loricrin and connexin gene mutations were absent in one case. 255 It differs from EKV by a greater incidence of palmoplantar keratoderma and the absence of migratory erythematous lesions. 256 As the name suggests, the symmetry of the lesions in this variant is more striking. 256 Profilaggrin N-terminal domains are aggregated with mutant loricrin within condensed nuclei. These nuclei persist in the cornified layer as parakeratosis. 257 The ichthyotic variant of Vohwinkel’s syndrome also has this abnormality. Progressive symmetric erythrokeratodermia could well be reclassified in the future as Vohwinkel’s syndrome or under the simplistic title of loricrin keratoderma (OMIM 604117). 258 Heterogeneous phenotypes of loricrin keratoderma may be the result of genetic heterogeneity of loricrin mutations. 259
EKV is said to be one of the most responsive genodermatoses to oral retinoids, although their use in children remains controversial. 260 Relapse usually occurs on cessation of the treatment. 256 Topical tretinoin 0.05% cream has also been used. Others have reported a lack of response to retinoids, but success with tazarotene gel. 261 Topical tacalcitol has also been used. 249 As for all keratotic diseases, emollients and keratolytics should be used first, or as adjuvants.
The findings are not distinctive. There is hyperkeratosis, irregular acanthosis, very mild papillomatosis in some biopsies, and a mild superficial perivascular infiltrate of lymphocytes. Dyskeratotic, grain-like cells may be seen in the lower stratum corneum. 262 There may be parakeratosis and some vacuolation in the lower horny cells and in the granular layer. 242
Harlequin ichthyosis (OMIM 242500) is a severe disorder of cornification. It is usually incompatible with extrauterine life and is of autosomal recessive inheritance.28. and 263. There appears to be genetic heterogeneity.264.265. and 266. It is characterized by thick, plate-like scales over the entire body with deep fissures. 267 Ectropion, eclabium, and flattened ears are other features of the disease. 268 It has been reported in association with hypothyroidism and juvenile rheumatoid arthritis in one patient. 269
The gene responsible has recently been identified as ABCA12, a member of the adenosine triphosphate-binding cassette (ABC) superfamily of active transporters.270. and 271. The gene maps to chromosome 2q34. 272 The ABCA12 protein is involved in the transportation of key lipids to the stratum corneum and the formation of lamellar granules.270. and 273. Now that DNA diagnosis is available, it allows for robust prenatal and preimplantation testing without the need for fetal skin biopsies.270. and 274. Prenatal skin biopsy was previously carried out at about 19 weeks.275. and 276. In countries where DNA testing is not readily available, a fetal skin biopsy is best done at around 23 weeks, because of pitfalls in making the diagnosis at an early stage. In some countries terminations of the pregnancy are only allowed until 20 or 21 weeks gestation. 268
Harlequin ichthyosis is a disorder of epidermal keratinization in which there are altered lamellar granules and a variation in the expression of keratin and filaggrin.265. and 277. Lipid levels may be increased in the stratum corneum. 278 In some cases, a defect of protein phosphatase has been demonstrated. 266 This enzyme may alter the processing of profilaggrin to filaggrin. 266
Patients who survive the neonatal period develop a severe exfoliative erythroderma consistent with non-bullous congenital ichthyosiform erythroderma (CIE). 279 Survival beyond the neonatal period has been enhanced by the use of oral retinoids. 272
There is massive hyperkeratosis in all biopsies. Some cases have parakeratosis with a thin or absent granular layer280 while others have had persistence of the granular layer. 278
Keratinization occurs much earlier in hair canals than in the interfollicular epidermis in fetal skin development. 268 Follicular changes occur at about 15 weeks gestation. Interfollicular changes can usually be seen at 19 weeks. 268 Terminal hair development is also more developed in the scalp than in vellus hair regions, so that the scalp is the best site to biopsy. 268 This information is of no relevance where DNA testing is available.
The stratum corneum is thickened and contains lipid and vacuolar inclusions. 282 Lamellar granules are abnormal or absent;265.283. and 284. instead, dense core granules are produced. 281 The marginal band (cellular envelope of cornified cells) is present at birth, in contrast to collodion baby in which it is absent at birth, but may develop later.285. and 286.
Follicular ichthyosis (ichthyosis follicularis – OMIM 308205) is a rare, distinctive form of ichthyosis in which the abnormal epidermal differentiation occurs mainly in hair follicles.287. and 288. Clinically there are spiny follicular papules. 289 Its onset is at birth or in early childhood. It is an X-linked recessive condition, but two female patients have now been reported. 290 The involved gene has not been characterized. The hyperkeratosis is more prominent on the head and neck. Photophobia and alopecia are often present, 291 leading to the eponym IFAP (ichthyosis follicularis, alopecia, atrichia, photophobia). 289 Three of the patients reported have also had acanthosis nigricans-like lesions. 287 The case reported as ‘ichthyosis cribriformis’ had keratotic cones and not spiny keratin excrescences, 292 but it may be a related condition.
A moderate response to retinoid therapy has been recorded. 293
There is marked follicular hyperkeratosis which is compact and extends deep within the follicle. There is a prominent granular layer. 287 In keratosis pilaris the hyperkeratosis has a more open basket-weave pattern and is confined to the infundibular region of the follicle.
Acquired ichthyosis, which occurs in adult life, is similar to ichthyosis vulgaris both clinically and histologically. 294 The subject was reviewed in 2006. 295 It is usually associated with an underlying malignant disease, particularly a lymphoma, 296 but it usually appears some time after other manifestations of the malignant process. Ichthyosis has also been associated with malnutrition, sympathectomy, 297 hypothyroidism, 298 leprosy, HIV infection, 299 HTLV-1 infection, 300 sarcoidosis,301. and 302. diabetes mellitus, 303 eosinophilic fasciitis304 and drugs such as clofazimine, pravastatin, 305 allopurinol, hydroxyurea,295. and 306. cimetidine, 295 fenofibrate, nicotinic acid, and nafoxidine. 307 Many of the drugs interfere with lipid metabolism. An ichthyosiform contact dermatitis may follow the repeated application of antiseptic solutions containing cetrimide. 308 There is compact orthokeratosis and/or parakeratosis without spongiosis. Cetrimide appears to act on the lipids and enzymes of the lamellar bodies. 308 Acquired ichthyosis is sometimes seen in the recipients of bone marrow transplants.309. and 310. Acquired ichthyosis must be distinguished from asteatosis (dry skin). It may be related to an essential fatty acid deficiency in some cases. 302
In acquired ichthyosis, there is compact orthohyperkeratosis with a reduced or absent granular layer. 295
Pityriasis rotunda, which is manifested by sharply demarcated, circular, scaly patches of variable diameter and number, is probably a variant of acquired ichthyosis. It is more common in black and oriental patients than in whites. An underlying malignant neoplasm or systemic illness is often present.311.312.313.314.315. and 316. Pityriasis rotunda may also occur as a familial disease, suggesting that there are two types with significant prognostic differences.317.318. and 319. It is particularly common on the island of Sardinia, where familial cases are not uncommon, suggesting that it is a genodermatosis. 320 Immunohistochemical studies have shown a marked reduction in filaggrin and loricrin expression in lesional skin and a reduction, on light microscopy, 321 in keratohyaline granules, beneath a layer of compact orthokeratosis. 321
Refsum’s disease (OMIM 266500), a rare autosomal recessive disorder, is characterized by ichthyosis, cerebellar ataxia, peripheral neuropathy, and retinitis pigmentosa. 28 The skin most resembles ichthyosis vulgaris but the onset of scale is often delayed until adulthood. There is an inability to oxidize phytanic acid, and improvement occurs when the patient adheres to a diet free from chlorophyll, which contains phytol, the precursor of this fatty acid. 28 It is caused by a mutation in the gene encoding phytanoyl-CoA hydroxylase (PAHX, or PHYH) at 10pter–p11.2, or the gene encoding peroxin-7 (PEX7) at 6q22–q24. 322 Decreased phytannic acid oxidation is also observed in cells lacking PEX7, so the two genes have a related function.
A biopsy will show hyperkeratosis, a granular layer that may be increased or decreased in amount, and some acanthosis. Basal keratinocytes are vacuolated and these stain for neutral lipid. 323 Lipid vacuoles are also present in keratinocytes in the rare Dorfman–Chanarin syndrome in which congenital ichthyosis is present (see below). 324
There are non-membrane-bound vacuoles in the basal and suprabasal keratinocytes. 323
There are a number of rare syndromes in which ichthyosis is a feature. As their histopathology resembles one of the already described forms of ichthyosis, they will be discussed only briefly.
The Sjögren–Larsson syndrome (OMIM 270200) is an autosomal recessive neurocutaneous disorder characterized by the triad of congenital, pruritic ichthyosis (most resembling lamellar ichthyosis), spastic paralysis, and mental retardation.325.326. and 327. Other changes include short stature, kyphoscoliosis, photophobia, and a reduction in visual acuity. 328 The Mongolian spot reported in one patient appears to have been coincidental. 329 Sometimes the ichthyosis does not develop until the first few months of life. An enzymatic defect in fatty alcohol oxidation has been identified.330. and 331. This is due to a mutation in the ALDH3A2 gene which codes for fatty aldehyde dehydrogenase. 327 Over 70 mutations have been described, including one leading to partial reduction in enzyme activity.332.333.334. and 335. A prenatal diagnosis can be made by fetal skin biopsy336 or biochemical analysis of a cultured chorionic villus. 337 The thickened keratin layer may still retain its basket-weave appearance. 338 At other times it is compact. There is also acanthosis, mild papillomatosis and a thickened granular layer. Abnormal lamellar inclusions are present in the cytoplasm of granular and horny cells by light and electron microscopy.327. and 328.
The ‘KID’ syndrome (OMIM 148210) comprises keratitis, ichthyosis, and deafness.340.341.342.343.344.345.346.347.348.349.350. and 351. It is rare with fewer than 100 cases reported. It is due to a mutation in the GJB2 (CX26) gene which encodes the gap-junction protein connexin 26.352.353.354.355. and 356. It maps to chromosome 13q12.11. It is really an ectodermal dysplasia357 and the ‘ichthyosis’ is related to erythrokeratodermia. 358 Scarring alopecia, palmoplantar keratoderma, 359 and susceptibility to infection360 are other clinical manifestations. 361 Trichothiodystrophy-like hair abnormalities have been reported in this condition. 362 Mutations in the connexin 26 gene are also associated with Vohwinkel syndrome and with sensorineural hearing loss. Various tumors, including tumors of the external root sheath, may occur in KID syndrome.363. and 364. Squamous cell carcinoma of the skin can also occur in the ‘HID’ syndrome (OMIM 602540), another autosomal dominant disease characterized by sensorineural deafness and spiky hyperkeratosis affecting the entire skin. 365 The ichthyosis in ‘HID’ syndrome was originally said to be of epidermolytic hyperkeratotic type (ichthyosis hystrix), giving rise to the eponym HID (hystrix ichthyosis and deafness). 330 A recent paper makes no mention of this finding but states that the electron microscopic findings are different. 365 The two syndromes are allelic.
Information on the autosomal recessive form (OMIM 242150) is scanty.
Treatment with oral and/or topical acitretin has shown promising results. 366
The Conradi–Hünermann–Happle syndrome (OMIM 302960) combines punctate chondrodysplasia with ichthyosiform lesions that may be diffuse, linear following Blaschko’s lines,367.368. and 369. or erythrodermic.370.371. and 372. Alopecia, nail changes, and cataracts are other manifestations. It is an X-linked dominant form of chondrodysplasia punctata due to mutations in the EBP (emopamil binding protein) gene located at Xp11.23–11.22. 373 This gene plays a role in cholesterol biosynthesis.368. and 374. A block in the same pathway occurs in the Smith–Lemli–Opitz syndrome (OMIM 270400), which also has chondrodysplasia. 373 Numerous different mutations have been described, but no clear genotype–phenotype correlation has emerged.375. and 376.
Mild ichthyosiform changes have been reported in two other variants of chondrodysplasia punctata. 372 The X-linked recessive form (OMIM 320950) is due to mutations in the arylsulfatase (ARSE) gene at Xp22.3 and the autosomal recessive form (OMIM 215100) is due to mutations in the PEX7 gene encoding the peroxisome targeting signal 2 receptor on 6q24–q22. This is the same gene reported to cause one type of Refsum’s disease, raising the likelihood that they are the different phenotypic expressions of the same disease, rather than two allelic conditions.
A skin biopsy shows hyperkeratosis, a prominent granular layer, dilated ostia of pilosebaceous follicles with keratotic plugging, dilated acrosyringia, and calcium in the stratum corneum.377. and 378. Intracorneal calcification, often localized to keratotic follicular plugs, is diagnostic of this condition. 372 Parakeratosis with a diminished granular layer sometimes occurs. 371 Dyskeratotic cells may be present in the hair follicles. 368
The Neu–Laxova syndrome (OMIM 256520) is a rare, lethal, autosomal recessive ichthyosis characterized by the presence of intrauterine growth retardation, microcephaly with abnormal brain development, ichthyosis, and edema.379. and 380. A thick membrane is usually present at birth, another example of a ‘collodion baby’. 381 No gene mutations have been detected.
The ‘CHILD’ syndrome (OMIM 308050) is an acronym for congenital hemidysplasia, ichthyosiform erythroderma (nevus) and limb defects.27.382.383.384.385.386. and 387. The ‘nevus’ takes the form of an erythematous plaque with a sharp border and yellowish, waxlike scaling. 388 This is a marked affinity for body folds. As in Conradi–Hünermann–Happle syndrome, there is also an abnormality in peroxisomal function, indicating the close relationship of these two conditions.368. and 371. It is an X-linked dominant disorder that is lethal in males.100. and 389. It is due to mutations in the NSDHL gene on chromosome Xq28.388. and 390. The gene plays an important role in cholesterol biosynthesis. Mild expression of the phenotype is sometimes present. 391 Variations include linear lesions, lesions along Blaschko’s lines, and verruciform xanthomas.100. and 392. Histopathological examination reveals psoriasiform epidermal hyperplasia and, sometimes, verruciform xanthoma change. 388 Squamous cell carcinoma has arisen in the affected ichthyosiform skin. 393 Electron microscopy in one case showed abnormal lamellar granules in the stratum corneum and upper prickle cell layer. 394
‘IBIDS’ (ichthyosis with trichothiodystrophy – OMIM 601675) combines ichthyosis with brittle hair, impaired intelligence, decreased fertility, and short stature.395. and 396. ‘PIBIDS’ is used when photosensitivity is also present. 397 It is associated with the defective repair of ultraviolet-induced DNA damage, as seen in xeroderma pigmentation, but skin neoplasms have not been seen until recently. 398 The brittle hair results from trichothiodystrophy. It is caused by defects in the ERCC2 gene, and less often the ERCC3 gene.
‘ILVASC’ (OMIM 607626) is the eponym for ichthyosis, leukocyte vacuoles, alopecia, and sclerosing cholangitis. It is caused by mutations in the claudin 1 (CLDN1) gene located at 3q28–q29. 390 Claudin 1 is a tight junction protein. It shares some clinical features with Dorfman–Chanarin syndrome (neutral lipid storage disease) – see below.
Multiple sulfatase deficiency (OMIM 272200) includes severe neurodegenerative disease, similar to metachromatic leukodystrophy, ichthyosis, and signs of mucopolysaccharidosis.28. and 399. It is an extremely rare autosomal recessive disorder affecting the activity of many sulfatases (arylsulfatase A, steroid sulfatase, and several mucopolysaccharide sulfatases), an explanation for the various clinical manifestations of this disease. 400 It is due to mutations in the sulfatase modifying function 1 (SUMF-1) gene at 3p26–p25. 390 The ichthyosis, not surprisingly, has some features of X-linked ichthyosis. If the ichthyosis is mild, treatment with emollients will suffice. 401
The ‘MAUIE’ syndrome consists of micropinnae, alopecia universalis, congenital ichthyosis, and ectropion. The early development of skin cancer has been reported in this syndrome. 402 It is not listed on the OMIM database.
Neutral lipid storage disease (Dorfman–Chanarin syndrome – OMIM 275630), in which neutral lipid accumulates in the cytoplasm of many cells of the body, combines fatty liver with muscular dystrophy and non-bullous ichthyosiform erythroderma.28.403. and 404. Other patterns of ichthyosis have been described.405. and 406. It is an autosomal recessive disorder in which a defect of acylglycerol recycling from triacylglycerol to phospholipid has been identified in fibroblasts. 407 It is due to mutations in the CGI-58 (ABHD5) gene located on 3p21.408.409. and 410. An excess of triacylglycerol accumulates in most cells. 411 Lipid droplets are found within leukocytes (Jordans’ anomaly). A skin biopsy shows hyperkeratosis, mild acanthosis, and discrete vacuolation of basal keratinocytes, sweat glands, and sweat ducts. 412 The vacuoles contain lipid.
Shwachman syndrome (Shwachman–Diamond syndrome – OMIM 260400) combines pancreatic insufficiency and bone marrow dysfunction with xerosis and/or ichthyosis.385. and 413. It is due to mutations in the SBDS gene at 7q11.
Other named and unnamed associations have been described.27.414.415. and 416. One of these is ichthyosis associated with the ARC syndrome (arthrogryposis, renal tubular dysfunction, and cholestasis – OMIM 208085) due to a mutation in the VPS33B gene on chromosome 15q26.1.417. and 418. Defective lamellar granule secretion has been described recently in this condition. 418 Another is the association of ichthyosis, follicular atrophoderma, hypotrichosis, and woolly hair (OMIM 602400). 419
In addition to the group of disorders usually categorized as the palmoplantar keratodermas, there are several rare genodermatoses that are usually regarded as discrete entities in which palmoplantar keratoderma is a major clinicopathological feature. These disorders include hidrotic ectodermal dysplasia (see p. 276), acrokeratoelastoidosis (see p. 261), pachyonychia congenita (see p. 261), tyrosinosis (see p. 260), and pachydermoperiostosis (see p. 312). Keratoderma may also occur as a manifestation of certain inflammatory dermatoses, such as pityriasis rubra pilaris, Reiter’s disease, psoriasis, Darier’s disease, and as a paraneoplastic manifestation. 420 These conditions will not be considered further in this chapter.
Palmoplantar keratoderma may result from mutations in keratin genes (pachyonychia congenita, epidermolytic hyperkeratosis of palms and soles) or in the genes regulating the desmosomal cadherins (plakophilin 1, plakoglobin, desmoplakin, desmoglein 1, and desmocollin 3). 421
The palmoplantar keratodermas are a heterogeneous group of congenital and acquired disorders of keratinization, characterized by diffuse or localized hyperkeratosis of the palms and soles, sometimes accompanied by other ectodermal abnormalities.422. and 423. Another classification system has also been proposed: the four major categories include diffuse, focal/circumscribed, and punctate palmoplantar keratodermas, and the palmoplantar ectodermal dysplasias. More than 20 subtypes have now been described in this latter category. 424 In one Indian study, the focal/circumscribed group was the most common type of palmoplantar keratoderma. 425 Categorization has been made on the basis of their mode of inheritance, sites of involvement, and associated abnormalities. 426 The autosomal recessive types are usually the most severe and include mal de Meleda, Papillon–Lefèvre syndrome, some mutilating variants, and a variant associated with generalized ichthyosis. 427 The other hereditary forms are autosomal dominant, although sporadic cases of most syndromes occur. 428
Another feature used to distinguish the various subtypes of keratoderma is the presence of hyperkeratosis beyond the palms and soles. These ‘transgrediens’ lesions occur in the Olmsted, Greither, Vohwinkel, and mal de Meleda types. 429 Onset of most keratodermas is at birth or in early infancy, but later onset is seen in the punctate and acquired forms. New syndromes continue to be reported or recognized.430. and 431. In one of these, palmoplantar keratoderma is associated with large ears, hypopigmented hair, and frontal skull bossing. 432 In another type, the Bothnian type (OMIM 600231), found in northern Sweden, the gene maps to 12q11–q13. 390 In yet another, a non-epidermolytic palmoplantar keratoderma is associated with sensorineural deafness and a mutation in the mitochondrial genome (mtDNA).433. and 434. There are at least two genotypic variants of palmoplantar keratoderma with deafness. One is due to a mutation in the connexin 26 gene (GJB2) at 13q11–q12 (OMIM 148350) and the other is due to the mitochondrial mutation just mentioned. It has consistently involved an A7445G point mutation (OMIM *590080). 434 In palmoplantar keratoderma with anogenital leukokeratosis, an absence of K6 and K16 has been detected. 435 No keratin or genetic studies were done on the case with oral leukokeratosis and recurring cutaneous horns of the lip. 436 In the Schöpf–Schulz–Passarge syndrome (OMIM 224750) there are associated apocrine hidrocystomas, hypodontia, hypotrichosis, and hypoplastic nails.437. and 438.
Brief mention will be made of the important clinical features of the various keratodermas.
Unna–Thost syndrome (OMIM 600962) usually presents in the first few months of life.439. and 440. Late onset has been recorded.425. and 441. Deafness is sometimes present, 442 while acrocyanosis and total anomalous pulmonary venous drainage are rare associations.443. and 444. Atopic dermatitis is another association. 445 Verrucous carcinoma has been reported in one case. 446 The syndrome may not be as common as once thought, because of the inclusion of cases of Vörner’s syndrome with which it is clinically, but not histologically, identical. 447 This epidermolytic hyperkeratotic type (OMIM 144200) is best kept separate from other cases. Two variants of Unna–Thost syndrome have been reported. One involves KRT1, which maps to the type II keratin cluster on chromosome 12q13, 448 and the other involves KRT16 on 17q12–q21.
In this ‘transgrediens’ form (hyperkeratosis beyond the palms and soles), the elbows and knees may be more involved than the palms and soles.422. and 449. It also involves the skin over the Achilles tendon. 450 It is associated with hyperhidrosis. The inheritance is autosomal dominant with reduced penetrance. 451 The gene has not been identified, but families have been described in whom the same dominant missense mutation gave rise to the amino acid change NI88S in keratin 1. 450 Histological changes are non-specific with hyperkeratosis and acanthosis. 452
Periorificial keratoderma and oral leukokeratosis accompany the transgrediens palmoplantar keratoderma, which is often mutilating.429.453.454.455.456.457.458. and 459. Alopecia and perianal keratotic plaques are sometimes present.460. and 461. This syndrome has been reported in twins. 462 Most cases are sporadic. 463 A closely related entity with corneal epithelial dysplasia has been reported. 464 Abnormal expression of keratins 5 and 14 appears to be the underlying disorder; 465 this remains to be confirmed. Plantar squamous cell carcinomas sometimes develop. 466
The histopathological findings on palmar skin include psoriasiform hyperplasia, hypogranulosis, and alternating parakeratosis and orthohyperkeratosis. 460
Vohwinkel’s syndrome is a family of genodermatoses which exhibits clinical and genetic heterogeneity. 469 There is a variant with deafness (OMIM 124500) and another with ichthyosis (OMIM 604117). In both forms there is a honeycombing pattern of keratoderma with starfish-shaped keratoses on the dorsa of the digits and linear keratoses on the knees and elbows.470.471.472. and 473. Ainhum-like constriction bands develop, leading to gangrene of the digits in adolescence.471.474.475. and 476. A recessive variant is associated with ectodermal dysplasia. 477 Congenital deaf-mutism is a rare association. 478 The subset of patients with mutilating keratoderma and sensorineural hearing loss (OMIM 124500) has a mutation in the GJB2 gene on chromosome 13q11–q12, which encodes the gap junction protein connexin 26. 479 The subset of patients with Vohwinkel’s keratoderma and an associated ichthyosiform dermatosis (OMIM 604117) has a mutation in the loricrin gene (LOR) on chromosome 1q21.469.480. and 481. The mutant loricrin is translocated into the nucleus of the keratinocyte and not into the cornified cell envelope as might be expected. 254 This form of the disease is now known as loricrin keratoderma. 482 It can be summarized as ‘honeycomb palmoplantar keratoderma with ichthyosis’. 482
The term ‘epidermolytic palmoplantar keratoderma’ (OMIM 144200) is widely used for this variant, reflecting the histological changes.483.484.485.486.487.488.489. and 490. It is clinically indistinguishable from the Unna–Thost variant. It has early onset and thick yellowish hyperkeratosis often with an erythematous border.491. and 492. A kindred with associated internal malignancy has been reported. 493 Mutations of keratin 9 (KRT9), which is specifically found in the suprabasal keratinocytes of palmoplantar epidermis and outer root sheath epithelium, have been found in this variant.491.494.495.496.497.498.499.500.501.502.503.504.505. and 506. At least 15 mutations in this gene have been reported.507. and 508. It is linked to chromosome 17q12–q21. 509 Mutations in the keratin 1 (KRT1) gene are found in epidermolytic hyperkeratosis (bullous ichthyosiform erythroderma – OMIM 113800), which may also be associated with a palmoplantar keratoderma (see p. 252). 510 Other kindreds with mutations in KRT1 have presented with only palmoplantar keratoderma and no generalized disease but in these cases the hyperkeratosis has either extended onto the proximal wrist flexure511 or involved other sites focally.142. and 192. This is the PS-1 variant of epidermolytic hyperkeratosis. It responds poorly to retinoids. 142
Epidermolytic palmoplantar keratoderma has occurred in several members of a family with Ehlers–Danlos syndrome, type III. 512 The genes for these two conditions are not closely linked. Focal variants have been described with the histology of epidermolytic hyperkeratosis. In one such case (OMIM 148730), focal palmoplantar keratoderma was associated with leukokeratosis. 513 Another variant is the unilateral palmoplantar verrucous nevus which represents mosaicism for a keratin 16 mutation. 514
Howel–Evans syndrome (tylosis with esophageal cancer – OMIM 148500) comprises palmoplantar keratoderma and esophageal cancer. The palmoplantar keratoderma begins early in life and affected family members develop a squamous cell carcinoma of the esophagus in middle adult life. 515 The gene has been linked to chromosome 17q25, which is distal to the type I keratin gene cluster. 424 Several candidate genes have been suggested including envoplakin390 and DMC1. Until identified and fully characterized, the gene for Howel–Evans syndrome has been called TOC. Some authors distinguish two types (A and B) depending on the age of onset. 516 Squamous cell carcinoma sometimes develops in the thickened skin of the palms. 517 Interestingly, palmoplantar keratoderma has been reported in one patient with postcorrosive stricture of the esophagus. 518
In Papillon–Lefèvre syndrome (OMIM 245000), an autosomal recessive condition with a prevalence of 1–4 per million, there is periodontosis accompanied by premature loss of the deciduous and permanent teeth.519.520.521.522. and 523. It is a type IV palmoplantar keratoderma (along with the Haim–Munk syndrome). They differ from the other three types by the presence of early-onset periodontitis and their autosomal recessive inheritance. 524 Involvement of the elbows and knees and calcification of the choroid plexus may occur.525. and 526. It has been associated with pseudoainhum of the thumb. 527
This condition is caused by a mutation in the cathepsin C gene (CTSC) located at chromosome 11q14.1–q14.3.528.529.530. and 531. The phenotypically related Haim–Munk syndrome is an allelic mutation. 532 There may be mild phenotypic expression of the disease with late onset and mild skin or periodontal disease. 533 In one late-onset case, no mutations in the cathepsin C gene were found, suggesting the possibility of another genetic cause. 534 The severity of the periodontal disease does not correlate with the severity of the skin lesions. 535 This syndrome has been reported in two families with oculocutaneous albinism. 536 The genes for these two conditions are closely situated on chromosome 11q14. 536
Periodontitis is also present in the ‘HOPP’ syndrome (hypotrichosis, acro-osteolysis, palmoplantar keratoderma, periodontitis – OMIM 607658) but there is no mutation in the cathepsin C gene. 537 The original report described the keratoderma as being striate. 538
The Haim–Munk syndrome (OMIM 245010), which is allelic to the Papillon–Lefèvre syndrome, is an extremely rare autosomal recessive condition caused by mutations in the cathepsin C gene (CTSC) located at chromosome 11q14.1–q14.3. 531 It is characterized by palmoplantar hyperkeratosis, severe early-onset periodontitis, pes planus, arachnodactyly, and acro-osteolysis.524. and 531.
Mal de Meleda (OMIM 248300), a rare, autosomal recessive variant of palmoplantar keratoderma, was first described in families living on the small island of Meleda in the Adriatic Sea.541. and 542. The responsible gene, ARS B, which encodes SLURP-1 (secreted mammalian Ly-6/uPAR related protein-1), is localized to 8qter.543.544.545.546. and 547. Onset is at birth or in infancy. Lesions may extend from the palms and soles onto the dorsum of the hands and feet respectively, although there is a sharp ‘cut-off’ at the wrists and ankles (transgrediens type).541.548.549.550. and 551. Pseudoainhum is a rare complication. 552 Hyperhidrosis leads to severe malodorous maceration. 426Palmoplantar keratoderma of Sybert is clinically similar but its inheritance is autosomal dominant; only one family has been reported. 447Nagashima-type palmoplantar keratoderma, reported from Japan, is an autosomal recessive, transgressive, and non-progressive palmoplantar keratoderma.553. and 554. Although similar to mal de Meleda, there were no mutations in the SLURP-1 gene. 554
Carvajal syndrome (OMIM 605676) has similar phenotypic features to Naxos disease. It is caused by mutations in the gene encoding desmoplakin (DSP), which maps to 6p24. 557 It is characterized by palmoplantar keratoderma and woolly hair, but the accompanying cardiomyopathy is of dilated type. A similar mutation is seen in type II striate palmoplantar keratoderma (see below).
Two Swedish families have been reported with very thick hyperkeratotic plaques on the dorsal aspect of the fingers. 447
This syndrome (OMIM 610644) is an extremely rare autosomal disease characterized by palmoplantar keratoderma, sclerodactyly, nail anomalies, and squamous cell carcinomas of affected skin.447.558.559. and 560. Additional features include dental anomalies, hypogenitalism, telangiectasia of the lips, flexor contractures of the little finger, and poikiloderma-like lesions on the nose.561. and 562. There is a mutation in the gene (RSPO1) encoding R-spondin-1. The related R-spondin gene RSPO4 produces autosomal recessive congenital anonychia.563.564. and 565.
Punctate palmoplantar keratoderma (OMIM 148600) is a rare, localized, hereditary variant of palmoplantar keratoderma characterized by discrete, hard, keratotic plugs arising in normal skin.566. and 567. A subset with verrucoid lesions has been reported. 568 Another variant is confined to the palmar and digital creases. 569 It is more common in black people.570.571.572. and 573. The punctate keratoses coalesce into a more diffuse pattern over the pressure points on the soles.574. and 575. The plugs form again if removed. Wood’s light excites white fluorescence in lesions of this condition. 576 Onset is usually in adolescence, but it may be much later. 577 An underlying malignancy has been present or developed subsequently in a few cases.578. and 579. Widespread lentigo simplex was present in one case. 580 Punctate lesions, which are characterized histologically by a cornoid lamella, are best classified as punctate porokeratotic keratoderma; 581 they are clinically indistinguishable from the other punctate forms.568.571.572.573.578.582. and 583. The term ‘spiny keratoderma’ has also been used for this type.584. and 585. Cole disease, in which hypopigmentation accompanies the punctuate keratoderma, is probably a variant. 586 The molecular basis of this disease is unknown. The keratin gene clusters on chromosomes 12 and 17 have been excluded as the causative genes. In 2005 a gene, localized to 15q22.2–q22.31, was identified in a four-generation Chinese family. 574
Topical agents such as keratolytic ointments, retinoids, and vitamin D analogues result in minor improvement in the condition. Combination therapy with 40% salicylic acid ointment and oral acitretin produced good results in one patient. 580 Acitretin alone has also been used. 587 Other therapies used include mechanical debridement, salicylic acid, topical tazarotene gel, topical fluorouracil, and surgical excision. 588
Striate palmoplantar keratoderma is a very rare form of palmoplantar keratoderma characterized by linear hyperkeratotic streaks along the volar surface of the fingers and focal keratoderma over the soles. It is a form of circumscribed keratoderma (see below). It has been reported in monozygotic twins. 589 Sporadic cases also occur. 590 Three types have been identified:
Both desmoglein 1 and desmoplakin are critical components of the desmosomal plaque and associated keratin intermediate filaments in the upper epidermis.556. and 593. Mutations of the tail domain of keratin 1 have been shown to affect the function of the plaque during cornification. 592 Disadhesion and partial acantholysis of keratinocytes in the spinous and granular cell layers have been reported in patients with DSG1 mutations. 594
Focal areas of thickening, sometimes tender, may develop on the palms and soles. 595 Some have an autosomal recessive inheritance. This is a clinically heterogeneous group, 595 which includes the conditions of hereditary painful callosities (keratosis palmoplantaris nummularis)596.597. and 598. and keratoderma palmoplantaris striata (striate palmoplantar keratoderma).599.600. and 601. This condition is considered above. Corneal dystrophy has been present in some circumscribed variants. 595 Localized hypertrophy of the skin of the soles and palms can occur in the Proteus syndrome, a rare disorder in which the major manifestations are skeletal overgrowth, digital hypertrophy, exostoses of the skull, subcutaneous lipomas, and, sometimes, epidermal nevi. 602 Tyrosinemia type II (Richner–Hanhart syndrome) also has localized lesions (see below). Focal palmoplantar callosities have been reported in a patient with non-Herlitz junctional epidermolysis bullosa. 603 All of these circumscribed variants have been classified as ‘nummular hereditary palmoplantar keratodermas’, 447 a term which appears unsuitable with regard to the morphological diversity of the lesions. A variant of epidermolytic palmoplantar keratoderma, localized to the palm and sole of one side of the body and following Blaschko’s lines, has been reported. The term ‘unilateral palmoplantar verrucous nevus’ was suggested for this mosaic mutation in KRT16. 514 Its histology was that of epidermolytic hyperkeratosis.
Palmoplantar keratoderma or discrete keratotic lesions may rarely develop in cases of myxedema,604.605. and 606. psoriasis, 607 lichen planus, pityriasis rubra pilaris, 608 mycosis fungoides, 609 lymphoma, 610 multiple myeloma, 611 and internal cancers, 612 following exposure to arsenic and after the menopause or following bilateral oophorectomy (keratoderma climactericum). 613 It has also been reported in a patient with a pemphigus-like immunobullous disease associated with antibodies to desmocollin 3. 421 Rugose lesions of the palms may occur in association with acanthosis nigricans, so-called ‘tripe palms’ (pachydermatoglyphy);614.615. and 616. in a few instances, this condition has been reported in association with a cancer, without concurrent acanthosis nigricans.617. and 618. TGF-α has been incriminated in the pathogenesis of this paraneoplastic change. 619 Keratoderma has been described in patients with AIDS, following the infusion of glucan, which was used as an immunostimulant. 620 Palmoplantar keratoderma has also followed the use of tegafur, in the treatment of an adenocarcinoma of the colon621 and one of the gallbladder. 622 There was a preceding acral erythema. Similar side effects have been reported with fluorouracil, a closely related drug. 621 Exposure to herbicides was implicated in another case. 623 Acquired punctate lesions on the palms and soles may follow dioxin intoxication. 624 Other drugs that may cause this condition include imatinib, 625 capecitabine, 626 and venlafaxine. 627
Finally, mention will be made of a type of callus that is a computer-related occupational dermatosis resulting from friction and pressure on the hand secondary to computer mouse use. It has been called ‘mousing callus’. 628
Aquagenic acrokeratoderma (transient reactive papulotranslucent acrokeratoderma, aquagenic palmoplantar keratoderma, aquagenic syringeal acrokeratoderma) was described by English and McCollough in 1996. 629 It is characterized by the rapid development of transient whitish, usually symmetric, hypopigmented flat-topped papules and plaques on palmar or plantar630 skin after exposure to water or sweating.629.631.632.633.634. and 635. Sometimes lesions develop on the dorsal aspect of the fingers. 636 Unilateral cases have been reported.637. and 638. Eccrine duct prominence is sometimes observed in these lesions, which arise within a few minutes after immersion (‘hands-in-the-bucket’ sign). 639 They may be asymptomatic or accompanied by a pruritic or burning sensation. 640 They resolve after variable drying periods. 641
It involves predominantly young females, 642 but cases in males have been reported.639.640.643. and 644. Familial cases have been recorded. 645 Most cases are transient and reactive.633. and 646. It has been reported in patients with cystic fibrosis, 640 cystic fibrosis carriers, 647 asthma, allergic rhinitis, congenital cardiomyopathy, 644 palmar erythema, hyperhidrosis, 630 melanoma, and after the ingestion of aspirin637 or rofecoxib. 648 Sodium retention in the skin may be a mechanism in some cases. 648 Reduced activity of transglutaminase resulting in alterations in the cornified cell envelope and barrier function is another possible pathogenic mechanism.
Spontaneous amelioration occurs in some patients. Topical aluminum chloride or 12% aluminum lactate cream633 has produced a remarkable response in one week.635. and 642.
The cornified layer has a spongy appearance. 642 There is dilatation of the intraepidermal eccrine ducts and some hyperkeratosis around these dilated ducts. 639 A distinctive crenulated appearance has been reported in the eccrine coil. 649 There is aberrant staining for aquaporin 5 (AQP5) in the sweat glands, suggesting that the condition stems from dysregulation of sweating. 650
The rare interdigital variant is characterized by a symmetrical keratoderma localized to the interdigital spaces of the fingers. 651
The diffuse forms show prominent orthokeratotic hyperkeratosis, with variable amounts of focal parakeratosis. The granular layer is often thickened. There is also some acanthosis of the epidermis and a sparse, superficial, perivascular infiltrate of chronic inflammatory cells. In Greither’s syndrome there are circumscribed foci of orthokeratotic hyperkeratosis located on delled areas of the epidermis. 451 In a case of Olmsted’s syndrome reported recently, there was massive hyperkeratosis and a reduced to absent granular layer. 652 In the Huriez syndrome there is massive hyperkeratosis, marked acanthosis, and hypergranulosis. 653 The scleroatrophic areas show thickening of the dermal collagen bundles. 653
The epidermolytic form shows epidermolytic hyperkeratosis. This tissue reaction is described on page 264.
The punctate forms show a dense, homogeneous keratin plug which often results in an undulating appearance in the epidermis.577. and 582. There is usually a slight depression in the epidermis beneath the plug (Fig. 9.6). 582 Those punctate cases with a parakeratotic plug are best classified as punctate porokeratotic keratoderma (Fig. 9.7 and Fig. 9.8). 654 Focal acantholytic dyskeratosis was present in the punctate lesions in one reported case. 655
Punctate keratoderma. There is slight depression of the epidermis beneath a keratin plug. There is a pit in the adjacent stratum corneum. (H & E)
Punctate porokeratotic (spiny) keratoderma. A wide cornoid lamella is present towards one edge of the field. (H & E) (Photograph kindly supplied by Dr J.J. Sullivan.)
Punctate porokeratotic (spiny) keratoderma. The granular layer is absent beneath the parakeratotic zone. (H & E) (Photograph kindly supplied by Dr J.J. Sullivan.)
In hereditary callosities with blisters, there is intraepidermal vesiculation with cytolysis of keratinocytes and clumping of tonofilaments. 596 The changes resemble those seen in pachyonychia congenita (see p. 261). In other cases, epidermolytic hyperkeratosis has been present. 598
Ultrastructural studies have not shown consistent abnormalities. It seems that the normal association of filaggrin and keratin filaments does not occur in the stratum corneum. 422 Nucleolar hypertrophy has been noted in the punctate form. 582 Ultrastructural studies of the Papillon–Lefèvre syndrome have shown lipid-like vacuoles in corneocytes, abnormally shaped keratohyaline granules and a reduction in tonofilaments. 656 Vacuoles and many membrane-coating granules were seen in corneocytes in Vohwinkel’s syndrome. 473
Oculocutaneous tyrosinosis (OMIM 276600), also known as tyrosinemia II and the Richner–Hanhart syndrome, is an extremely rare, autosomal recessive genodermatosis caused by a deficiency of hepatic tyrosine aminotransferase (tyrosine transaminase).657.658.659.660.661. and 662. The gene is located at 16q22.1–q22.3. It is usually characterized by corneal ulcerations and painful keratotic lesions on the palms and soles, but ocular lesions have been absent in some kindreds. 663 The palmoplantar lesions vary from fine 1–2 mm keratoses to linear or diffuse keratotic thickenings.661. and 664. Erosions and blisters have also been reported.665.666. and 667. Mental retardation is often present. The condition is treated by a dietary restriction of tyrosine and phenylalanine. If started early, mental retardation may be avoided. 666
The palmoplantar lesions show prominent hyperkeratosis and parakeratosis with variable epidermal hyperplasia. Scattered mitoses and multinucleate keratinocytes may be present.668. and 669. Epidermolytic hyperkeratosis and intraepidermal bulla formation have been present in some families. 665
Acrokeratoelastoidosis (Costa’s papular acrokeratosis – OMIM 101850) 671 is a rare variant of palmoplantar keratoderma which occurs both sporadically and as an autosomal dominant genodermatosis.672.673. and 674. No gene has been implicated although early linkage studies suggested the ACP1 gene; another study suggested a gene on chromosome 2. 675 This has never been confirmed. There appears to be some variability in the morphological expression of the disease.676. and 677. Its onset is in childhood or early adult life and there is a female predominance. There are multiple, small (2–5 mm in diameter), firm, translucent, asymptomatic papules which are most numerous along the junction of the dorsal and palmar or plantar surfaces of the hands and feet respectively.678. and 679. Larger plaques also develop by coalescence of the papules. 680 They may be localized to the sides of the fingers, particularly the inner side of the thumb and the adjoining index finger. In this situation, they resemble clinically the lesions seen in collagenous and elastotic plaques of the hands (see p. 343), an acquired entity which is found predominantly in males over the age of 50 and which results from trauma and actinic damage. 671 In acrokeratoelastoidosis there may also be a mild diffuse hyperkeratosis of the palms and soles; this was a prominent feature in one report. 681 There may also be isolated lesions over interphalangeal joints and elsewhere on the dorsum of the hands and feet. 682 A variant of acrokeratoelastoidosis has been found on the palms in systemic scleroderma. 683
The treatment is a matter of debate. No curative treatment has been reported to date. 684 Options include topical keratolytics, retinoids, or corticosteroids. Systemic immunosuppressive therapy with prednisone or methotrexate has also been used. 684 Cryosurgery has been unsuccessful while the erbium:YAG laser has produced slight improvement. 684
There may be slight hyperkeratosis with a shallow depression in the underlying epidermis, which shows a prominent granular layer and mild acanthosis. The dermis may be normal or slightly thickened. The elastic fibers are decreased in number, thin and somewhat fragmented in the mid and deep reticular dermis. 686 In some cases the elastic fibers are coarse and fragmented. 682 Cases have been reported with no light microscopic changes in the dermis. 672 The group which lacks elastorrhexis has been designated ‘focal acral hyperkeratosis’.687.688. and 689. Their relationship is controversial, particularly as the epidermal changes of focal acral hyperkeratosis are caused by increased proliferation and differentiation of lesional keratinocytes. 690 The orthohyperkeratosis overlies a clavus-like depression of the epidermis and prominent hypergranulosis. 691
In acrokeratoelastoidosis there is disaggregation of elastic fibers with fragmentation of the microfibrils. 672 In one report the fibroblasts contained dense granules in the cytoplasm and there was an absence of extracellular fibers, leading to the hypothesis that there was a block in the synthesis of elastic fibers by the fibroblasts. 692
Pachyonychia congenita is a rare genodermatosis in which symmetrical, hard thickening of the nails of the fingers and toes is the most striking and consistent feature.693.694. and 695. Various other abnormalities of keratinization are usually present. 696 These include palmar and plantar keratoderma, follicular keratoses on the extensor surfaces of the knees and elbows, keratosis pilaris, blister formation on the feet and sometimes on the palms, callosities of the feet, leukokeratosis of the oral mucosa (often complicated by candidosis),697. and 698. hair abnormalities, and hyperhidrosis of the palms and soles. Acro-osteolysis is an uncommon complication. 699 These clinical features typify the so-called ‘type I’ (Jadassohn–Lewandowsky – OMIM 167200) cases.696. and 700. Patients with the type II variant (Jackson–Lawler type – OMIM 167210) have the addition of natal teeth (teeth erupted prior to birth) and multiple cutaneous cysts, either epidermal cysts or steatocystomas,701. and 702. but no oral leukokeratosis. 703 Woolly hair was present in one case. 704 In type III, the features of type I are accompanied by leukokeratosis of the cornea. Type IV combines the clinical features of the other types with laryngeal lesions, mental retardation, and alopecia. 705
There is some variability in the age of onset of the various manifestations, but nail changes are usually present at birth or soon afterwards and become progressively more disfiguring over the first year of life. Nail dystrophy of late onset (pachyonychia congenita tarda) has been reported.706.707.708. and 709.
Pachyonychia congenita is usually inherited as an autosomal dominant trait with variable expression and high penetrance, although several cases with autosomal recessive inheritance have been reported. 693Type II pachyonychia congenita is due to an abnormality in the keratin 17 (KRT17) gene and in its expression partner keratin 6b (KRT6B), 710 the former resulting from a gene mutation in the type I keratin cluster on chromosome 17q12–q21 and the latter from a mutation in KRT6B at 12q13. 711 Similar mutations in the KRT17 gene can cause steatocystoma multiplex with little or no nail dystrophy. 712Type I pachyonychia congenita results from a mutation in the helix initiation peptide of the keratin 16 (KRT16) gene713. and 714. or in the keratin 6a (KRT6A) gene located at 12q13.715.716. and 717. Mutations of KRT6A are said to be more frequent than those of KRT16. 718
The thickened nails and callosities have been treated with urea paste 40% with good results. 719 As plantar sweating at high ambient temperatures increases the blistering of the plantar callosities, control of the sweating with injections of botulinum toxin has been carried out. 720 This relieves the pain associated with walking on these callosities. 720
Mention is made here for completeness of ectopic nails. They have been reported on the volar surface of fingers and toes and elsewhere. 721 Ectopic plantar nails are smaller than normal ones. 722 Ectopic nails are most frequently caused by traumatic inoculation of nail matrix. They may be associated with polydactyly. 723 Congenital malalignment of the great toenails is another malformation. It is heritable. 724 The yellow nail syndrome (OMIM 153300) and other diseases of nails will not be considered further.725.726.727.728.729.730. and 731.
The involved mucous membranes and skin, including the nail bed, show marked intracellular edema involving cells in the upper malpighian layer and in the thickened stratum corneum.693.696.732.733.734. and 735. There is sometimes rupture of cell walls, particularly with lesions on plantar surfaces, with the formation of intraepidermal vesicles. The epidermis is markedly thickened as a consequence of the edema and the accompanying hyperkeratosis and focal parakeratosis.
The hyperkeratotic papules on the knees and elbows show hyperkeratosis, a prominent granular layer, and acanthosis.693. and 736. There is usually a thick horny plug extending above the infundibulum of the hair follicle. In one case, the plug resembled a cornoid lamella; 737 in another, the plug, which was not related to a follicle, penetrated into the dermis in the manner seen in Kyrle’s disease (see p. 273). 738 Horny plugs have also been described in sweat pores. 736
The plantar callosities show hyperkeratosis, focal parakeratosis, and sometimes mild papillomatosis and acanthosis. The granular layer is thick except for the area overlying any papillomatous foci. 696
Two kindreds have been reported with pigment incontinence and amyloid in the papillary dermis. 739
The cornoid lamella is a thin column of parakeratotic cells with an absent or decreased underlying granular zone and vacuolated or dyskeratotic cells in the spinous layer. It is the key histological feature of porokeratosis and its clinical variants, but like some of the other minor ‘tissue reaction patterns’ (see Ch. 1, p. 12) can be found as an incidental phenomenon in a range of inflammatory, hyperplastic, and neoplastic conditions of the skin. The cornoid lamella represents a localized area of faulty keratinization and is manifest clinically as a raised keratotic or thin thread-like border to an annular, gyrate, or linear lesion.
Cornoid lamellation has been regarded as a clonal disease740 and as a morphological expression of disordered epithelial metabolism. 741 Abnormal DNA ploidy and abnormalities of keratinocyte maturation have been demonstrated in the epidermis in some lesions of porokeratosis.742.743.744. and 745. Overexpression of p53 has been detected in the nuclei of keratinocytes in the basal layers of the epidermis beneath the cornoid lamella.746.747.748. and 749. This is accompanied by a reduction in mdm2 and p21.750. and 751. Gene expression profiling of porokeratosis has demonstrated similarities with psoriasis. 752 Both are hyperproliferative disorders of keratinocytes. 752 Both show increased connexin 30 expression. 753
Porokeratosis is a genodermatosis with many different clinical expressions. 754 Lesions may be solitary or numerous, inconspicuous or prominent, small or large (giant),755.756.757.758. and 759. atrophic or hyperkeratotic, 760 and asymptomatic or pruritic. 761 The lip, 762 mouth, 763 face,764.765.766. and 767. scalp, 768 nail fold, 769 penis,770. and 771. scrotum, 772 genitocrural and perianal regions,773.774.775.776. and 777. and natal cleft778 are sites that are rarely involved. Facial lesions may be destructive and disfiguring.779. and 780. Linear facial lesions have also been seen. 781 Involvement of a burn scar has been reported. 782 Other rare clinical associations include Crohn’s disease, 783 trisomy 16, 784 chronic renal failure,785. and 786. hepatitis C virus-related hepatocellular carcinoma, 787 cholangiocarcinoma, 788 previous renal transplant,789.790.791. and 792. bone marrow transplantation, 793 myelodysplastic syndrome, 794 lymphedema, 795 herpes simplex infection, 796 systemic lupus erythematosus, 797 microsatellite instability in association with hereditary non-polyposis colorectal cancer, 798 lesions localized to the access region for hemodialysis, 799 and onset after electron beam radiation,800.801. and 802. PUVA, 803 tanning salon use, 804 prolonged topical corticosteroid use, 805 and furosemide (frusemide). 806 The simultaneous development of DSAP and ovarian cancer in one patient may have been coincidental rather than a true paraneoplastic association. 807 Although it was originally regarded as a familial disease with autosomal dominant inheritance, 808 numerous non-familial cases have now been reported. Congenital cases are rare.781. and 809.
The most important clinical forms are porokeratosis of Mibelli (OMIM 175800), characterized by one or more round, oval or gyrate plaques with an atrophic center and a thin, elevated, guttered, keratotic rim which may show peripheral expansion, and disseminated superficial actinic porokeratosis (DSAP – OMIM 175900) consisting of multiple, annular, keratotic lesions less than 1 cm in diameter, with a hyperkeratotic, thread-like border and occurring particularly on the sun-exposed extremities.810.811. and 812. They usually develop during the third or fourth decade of life. DSAP appears to be heterogeneous with two forms identified – DSAP1 (OMIM 175900) tentatively linked to the 12q24.1 region, although nearby regions have also been suggested;813. and 814. and DSAP2 (OMIM 607728) on chromosome 15q25.1–26.1.815.816. and 817. Other candidate genes that have been suggested on chromosome 12 are slingshot 1 (SSH1) and ARPC3, both involved in the actin cytoskeleton pathway,817. and 818. and the SART3 gene. 819 A new locus has recently been discovered on 1p31.3–p31.1, for an autosomal dominant form of DSAP (DSAP3). 820
Rare clinical variants include a linear821.822.823.824.825.826.827.828.829. and 830. or systematized831. and 832. variant, a reticulate form, 833 a follicular variant, 834 an eruptive pruritic form, 835 porokeratosis plantaris discreta836. and 837. (painful plantar lesions), porokeratotic palmoplantar keratoderma discreta, 838 punctate porokeratotic keratoderma, also known as punctate porokeratosis,839.840.841.842.843. and 844. and porokeratosis punctata palmaris et plantaris – spiny keratoderma584.845. and 846. (PPPP – OMIM 175860 (asymptomatic pits or plugs, usually on the palms, digits, or soles and not universally accepted as a variant of porokeratosis)). 847 Other variants include linear palmoplantar porokeratotic hamartoma (the cornoid lamellae are not related to eccrine ostia), 848 porokeratosis plantaris, palmaris et disseminata849.850.851.852.853.854.855. and 856. (annular and serpiginous lesions on the palms and soles with later involvement of other areas (PPPD – OMIM 175850)), and the related superficial disseminated eruptive form.857. and 858. PPPD may be due to the same gene as DSAP1. 815 Another rare variant of porokeratosis is a hyperkeratotic verrucous type.859.860. and 861. In yet another variant craniosynostosis, anal anomalies, and porokeratosis are associated (CAP syndrome – OMIM 603116). 819 Closely related are the cases with cornoid lamellae in eccrine and hair follicle ostia, described as ‘porokeratotic eccrine ostial and dermal duct nevus’,862.863.864.865.866.867.868.869.870.871.872. and 873. ‘porokeratotic eccrine duct and hair follicle nevus’,874. and 875. and ‘reticular erythema with ostial porokeratosis’. 876 Porokeratotic eccrine ostial and dermal duct nevus is most commonly found on the palms and soles. It usually presents in early childhood with a linear distribution of asymptomatic papules. 873 The lesions may develop along Blaschko’s lines877. and 878. or be systematized. 879
Porokeratoma is a recently described solitary, hyperkeratotic plaque or nodule, sometimes verrucous, with a predilection for the distal extremities of middle-aged to elderly males. 880 It is akin to warty dyskeratoma and epidermolytic acanthoma. Histological examination shows a well-defined lesion, characterized by acanthosis and verrucous hyperplasia with prominent multiple and confluent cornoid lamellae. 880
The disseminated superficial actinic variant coexists rarely with other types of porokeratosis, particularly the linear type.823.827.881.882.883.884. and 885. The association of these two forms is an example of type 2 segmental involvement, occurring in an autosomal dominant skin disorder. 886 The coexistence of DSAP and porokeratosis of Mibelli has also been reported. 887 The various clinical types of porokeratosis are listed in Table 9.4.
DSAP may be exacerbated by exposure to ultraviolet light,888. and 889. but it does not appear to be due to radiation hypersensitivity. 890 There appears to be an inherent defect in the terminal differentiation program.890. and 891. Both DSAP and porokeratosis of Mibelli have developed in immunosuppressed patients.892.893.894.895.896.897.898. and 899. Age-related immunosuppression has been used to explain the occurrence of disseminated porokeratosis in the elderly. 900
Reflectance confocal microscopy has been used to diagnose DSAP, and to distinguish lesions from actinic keratoses. The cornoid lamella is easily identified by this technique. 901 Other dermoscopic methods can also be used.902. and 903. Fake sun tan lotion will highlight lesions of DSAP serendipitously. 904 Povidone-iodine can also be used to highlight the cornoid lamella. 905
Porokeratotic lesions are usually persistent although clearance with subsequent recurrence has been reported. 906 The development of squamous cell carcinoma748.885.907.908.909.910. and 911. or intraepidermal carcinoma912. and 913. is a rare clinical complication of several of the variants of porokeratosis. The linear type is particularly prone to malignant change.914.915. and 916. Fatal squamous cell carcinomas have developed in transplant-associated porokeratosis. 917 The term ‘malignant disseminated porokeratosis’ has been applied to several cases in which the porokeratotic lesions had a significant potential to undergo malignant degeneration.918. and 919. Allelic loss has been suggested as a possible pathogenetic mechanism for these tumors. 914
Treatment of porokeratosis is notoriously difficult. The author sees many cases (up to one per week) treated by surgical excision, but this seems an aggressive method of treatment for this condition. The clinical diagnosis has not been made in any of these cases. Shave excision, cryosurgery, electrocautery, carbon dioxide laser, topical 5-fluorouracil, keratolytics, and topical retinoids have also been used. 920 Successful treatment with 5% imiquimod cream has also been reported. 920 Fractional photothermolysis using an erbium-doped laser has also been effective. 921
It is important that the biopsy is taken across the edge of the peripheral rim in order to show the typical cornoid lamella, the features of which have been described above. Multiple cornoid lamellae will usually be seen in biopsies from the linear and reticulate form. There may be two cornoid lamellae, one on each side of a keratotic plug, at either edge of a lesion of DSAP (Fig. 9.9). 922 In porokeratosis of Mibelli there is invagination of the epidermis at the site of the cornoid lamella with adjacent mild papillomatosis. A PAS stain reveals that the cornoid lamella contains numerous purple granules, representing intracellular glycogen and glycoproteins. 923 Beneath the lamella there is absence or diminution of the granular layer, sometimes only several cells in width (Fig. 9.10). However, in the solitary palmar or plantar lesions the cornoid lamella is quite broad with a corresponding wide zone where the underlying granular zone is absent or markedly reduced. Sometimes one or more dyskeratotic cells are present in the spinous layer. Basal vacuolar change and vacuolated cells in the spinous layer are other changes found beneath the cornoid lamella. 924
Porokeratosis. There are two cornoid lamellae close together. One overlies an acrosyringium. Follicular infundibula may also be involved. (H & E)
Porokeratosis. The parakeratotic column (cornoid lamella) overlies an area several cells in width, in which the granular layer is absent. (H & E)
The case reported as multiple minute parakeratotic keratoses of the hand in a patient with systemic lupus erythematosus appears to be a variant of punctate porokeratotic keratoderma. 925
Beneath the lamella there are often dilated capillaries in the papillary dermis, associated with a lymphocytic infiltrate. A variant with numerous eosinophils resembling somewhat a bite reaction is exceedingly rare. The most prominent inflammatory changes are seen in DSAP, in which a superficial band-like infiltrate with lichenoid (interface) qualities is usually present. One should never sign out a case as lichen planus-like keratosis (benign lichenoid keratosis) until the presence of cornoid lamellae has been confidently excluded. The infiltrate is usually present inside the porokeratotic rim. 924 In addition, the epidermis between the lamellae is often atrophic in this form, with overlying hyperkeratosis, and the dermis may show solar elastotic changes. Focal epidermal necrosis and ulceration have been reported at the periphery of a lesion of porokeratosis.926. and 927. Subepidermal blistering is a rare event. 928 Amyloid has been found in the papillary dermis in several cases.929.930.931. and 932.
Scanning electron microscopy has shown bud-like spreading of the active edge, 933 while transmission electron microscopy has demonstrated that the basophilic granular material in the cornoid lamella consists of degenerate cells with pyknotic nuclei. 934 Langerhans cells are in close contact with degenerating keratinocytes. 935 The granular layer is inconspicuous with only small amounts of keratohyalin. Vacuolated cells and others showing filamentous degeneration (‘dyskeratotic’ cells) are often present. Apoptotic bodies may be seen in the basal layer and in the dermal papillae. 936
Epidermolytic hyperkeratosis is an abnormality of epidermal maturation characterized by compact hyperkeratosis, accompanied by granular and vacuolar degeneration of the cells of the spinous and granular layers (Fig. 9.11). It may be a congenital or an acquired defect. This histological pattern may be seen in a number of different clinical settings, some of which will be considered in other sections of this chapter. It may be:
• generalized: bullous ichthyosis (see p. 253)
• systematized or linear: epidermal nevus variant (see below and p. 669)
• palmoplantar: palmoplantar keratoderma variant (see p. 258)
• solitary: epidermolytic acanthoma (see below)
• multiple discrete: disseminated epidermolytic acanthoma (see below)
• incidental: focal epidermolytic hyperkeratosis (see below)
• solar keratosis related: a rare variant of solar keratosis (see p. 678)
• follicular: nevoid follicular epidermolytic hyperkeratosis (see below)
• mucosal: epidermolytic leukoplakia.
Epidermolytic hyperkeratosis. Compact orthokeratosis overlies an epidermis showing granular and vacuolar change in its upper layers. (H & E)
Epidermolytic hyperkeratosis is a relatively uncommon histological pattern in epidermal nevi, being present in only 8 of 160 cases reported from the Mayo Clinic (the clinical appearance of the lesions in this series is not described). 937 There are reports of cases with a systematized pattern (ichthyosis hystrix)938. and 939. and with a linear pattern (Fig. 9.12).939. and 940. Bullous ichthyosis (generalized epidermolytic hyperkeratosis) has been reported in the offspring of patients with the linear form of the disease.139. and 941. This is an example of the reverse of type 2 segmental involvement occurring in an autosomal dominant skin condition. It is unusual for those with segmental disease to pass on the generalized form.
Linear nevus of epidermolytic hyperkeratotic type. (H & E)
Focal epidermolytic hyperkeratosis has been found incidentally in a range of circumstances, including the wall of an infundibular cyst, 942 in a seborrheic keratosis, in a cutaneous horn and a skin tag, overlying an intradermal nevus and even in association with dermatoses. 194 Its incidence appears to be increased adjacent to dysplastic nevi.943. and 944. Epidermolytic hyperkeratosis has also been found rarely in solar keratoses, 945 and in leukoplakic lesions of the lips945. and 946. and prepuce. 947 It may involve the oral mucosa. 948 It has been observed on the labia and mons pubis, where it produced seven verrucoid papules resembling condylomas. 949 It may be found adjacent to any lesion as an incidental phenomenon.950. and 951.
The nevoid follicular variant952 presents as comedo-like follicular papules that may have the appearance of nevus comedonicus.953. and 954. A linear nevus comedonicus has been reported with the histological pattern of epidermolytic hyperkeratosis. 955 It may also involve the intraepidermal eccrine sweat duct.956. and 957.
If there is continuous involvement of the entire horizontal epidermis, then the patient is likely to have generalized epidermolytic hyperkeratosis (bullous congenital ichthyosiform erythroderma). The presence of focal epidermolytic hyperkeratosis with skip areas of normal-appearing epidermis correlates usually with mosaic disease. 958
There are similar ultrastructural changes in the different variants of epidermolytic hyperkeratosis. 959 There is clumping of tonofilaments and cytoplasmic vacuolation. Keratohyaline granules are of variable size.
Epidermolytic acanthoma is an uncommon lesion which presents clinically as a wart in patients of all ages.960. and 961. A disseminated form has also been reported;962.963.964. and 965. in one report of four cases, the lesions developed after sun exposure. 966 Trauma has also been incriminated. 965 Multiple scrotal epidermolytic acanthomas have been reported. They were thought to have resulted from the trauma of chronic scratching. 967
The lesions show the typical features of epidermolytic hyperkeratosis. The entire thickness of the epidermis may be involved or only the upper part of the nucleated epidermis (Fig. 9.13). There is a diminution in the expression of K1 and K10 in the altered granular layer of lesional skin. 961
Epidermolytic acanthoma. (A) Clinically this was a solitary, keratotic lesion. (B) Another case with a flatter epidermis and more overlying keratin. (H & E)
Acantholytic dyskeratosis is a histological reaction pattern characterized by suprabasilar clefting with acantholytic and dyskeratotic cells at all levels of the epidermis (Fig. 9.14). 968 It may also be regarded as a special subdivision of the vesiculobullous tissue reaction, but it is considered in this chapter because the vesiculation is not usually apparent clinically and the primary abnormality involves the tonofilament–desmosome complex with disordered epidermal maturation.
(A) Acantholytic dyskeratosis. (B) There is suprabasal clefting with occasional acantholytic and dyskeratotic cells. (H & E)
Like epidermolytic hyperkeratosis, acantholytic dyskeratosis may be found in a number of different clinical settings. 968 The two histological patterns have even been found in the same biopsy. 969 Acantholytic dyskeratosis may be:
• generalized: Darier’s disease (see p. 267)
• systematized or linear: zosteriform (segmental) Darier’s disease or linear nevus (see below and p. 669)
• transient: Grover’s disease (see p. 268)
• palmoplantar: a very rare form of keratoderma (see p. 260)
• solitary: warty dyskeratoma (see p. 271)
• incidental: focal acantholytic dyskeratosis (see below)
• solar keratosis related: acantholytic solar keratosis (see p. 139)
• mucosal: vulval and anal acantholytic dyskeratosis (see below).
The occurrence of acantholytic dyskeratosis in lesions with a linear or systematized distribution is best regarded as an example of segmental Darier’s disease induced by postzygotic mosaicism,970.971.972.973.974.975. and 976. and not as an example of epidermal nevus as previously thought.977.978.979. and 980. It is now well known that autosomal dominant skin disorders may sometimes become manifest in a mosaic form, involving the body in a linear, patchy, or circumscribed arrangement.981.982. and 983. The segmental disease usually shows the same degree of severity as that found in the corresponding non-mosaic trait. 981 Loss of heterozygosity for the same allele causes more severe changes.981. and 984. Segmental Darier’s disease shows the same mutations in the ATP2A2 gene that occur in generalized Darier’s disease. Unaffected skin does not show this mutation in keeping with mosaicism. 985 Acantholytic dyskeratosis is an uncommon finding in ‘epidermal nevi’, being present in only 2 of a series of 167 epidermal nevi reported from the Mayo Clinic. 937 Such lesions would have followed Blaschko’s lines, reflecting as it does genetic mosaicism. 986 The sole of the foot is a rare site for segmental disease. 987
Acantholytic dyskeratosis has also been reported as a rare pattern in familial dyskeratotic comedones, a condition with some features in common with nevus comedonicus (see p. 670). 988
Acantholytic dyskeratosis appears to affect cells within the germinative cellular pool of the epidermis. 989 The dyskeratosis that occurs within the acantholytic cells is probably a secondary phenomenon as the acantholytic cells are metabolically inert. 989
Although the term ‘focal acantholytic dyskeratosis’ is often used both for clinically inapparent incidental foci and for clinically apparent solitary lesions with the histological pattern of acantholytic dyskeratosis, 990 some authors restrict its use to its incidental finding in histological sections. This is not an uncommon event.991. and 992. Ko and colleagues have recently coined the term ‘acantholytic dyskeratotic acanthoma’ for this lesion, apparently on the basis that it is more than an incidental finding, and it often presents clinically as a basal cell carcinoma of the trunk. 993 The author sees at least one case each week of this phenomenon, whether it be incidental or otherwise. The term ‘papular acantholytic dyskeratoma’ has been applied to the clinically apparent solitary lesions, 994 and ‘papular acantholytic dyskeratosis’ to the exceedingly rare cases in which multiple lesions have developed on the vulva,995.996.997.998.999.1000. and 1001. perianal area,1002.1003. and 1004. or penis1005 (see below). A clinically apparent lesion has also been reported on the lip. 990 A case reported as ‘congenital acantholytic dyskeratotic dermatosis’ appears to be a variant of papular acantholytic dyskeratosis. There were multiple erosive papules and plaques located on the left thigh, left ankle, and right neck that were present at birth. 1006 There was no family history of Darier’s disease and no genetic studies were performed. 1006
Incidental focal acantholytic dyskeratosis is statistically increased in atypical melanocytic lesions. 1007
There is acantholytic dyskeratosis, as already defined (see above). Hyperkeratosis is less prominent in incidental lesions than in Darier’s disease. Warty dyskeratomas differ from focal acantholytic dyskeratomas by having more prominent villi, clefting, and corps ronds. Some of the genital and crural cases, mentioned above, have a histological resemblance to Hailey–Hailey disease, with marked acantholysis and little dyskeratosis. They belong to the recently recognized acantholytic subset of acantholytic dyskeratosis; another subset features dyskeratosis alone (see below).
In one case of papular acantholytic dyskeratosis of the anogenital region immunofluorescence showed intercellular IgG and C3 within the epidermis. 1008
Acantholysis, with little or no dyskeratosis, can be seen as an incidental phenomenon1009 or as a solitary tumor of the skin – acantholytic acanthoma (see p. 672).1010. and 1011. This pattern has also been found in multiple papules1012 and as a variant of epidermal nevus with horn-like processes. This latter case was reported as ‘nevus corniculatus’. 1013
A high proportion of the rare genital, crural, and perineal cases referred to as papular acantholytic dyskeratosis (see above) have had a histological resemblance to Hailey–Hailey disease, with prominent acantholysis and little or no dyskeratosis.997.1014.1015. and 1016. Other cases have resembled acantholytic dyskeratosis. 1001 An appropriate designation for these cases would seem to be ‘acantholytic dermatosis of the genitocrural/perineal region’. 1017 The exact classification of the vulval case with histological resemblance to pemphigus vegetans remains to be determined. 1018 Another case of this entity was reported as warty dyskeratoma.1019. and 1020. Incidentally, it responded to tazarotenic acid. 1019
Generalized acantholysis is seen in Hailey–Hailey disease. It is discussed on page 270. A peculiar form of acantholysis, localized to the acrosyringium, has been reported in several febrile patients. The clinical picture resembled Grover’s disease. 1021 The authors proposed the name ‘sudoriferous acrosyringeal acantholytic disease’. 1021
Finally, acantholysis with minimal dyskeratosis can be seen, as an incidental phenomenon, in other disease processes such as pityriasis rubra pilaris1022 and seborrheic keratoses.
Although regarded as a variant of epidermolytic acanthoma, the isolated lesion reported as ‘isolated dyskeratotic acanthoma’ is best classified here. 1023 It combined dyskeratotic cells throughout the epidermis with a parakeratotic horn containing large rounded cells at all levels. 1023
The term ‘acquired dyskeratotic acanthosis’ has recently been applied to a case in which multiple maculopapules, 3–8 mm in diameter, developed in sun-exposed areas. There were clusters of parakeratotic cells which appeared eosinophilic to ‘ghostlike’. The epidermis was papillomatous and acanthotic with foci of dyskeratotic keratinocytes. 1024
The proliferation of terms for solitary cases showing acantholysis, dyskeratosis or both, and in variable proportions, is bordering on the ridiculous. Perhaps prospective authors should attempt consolidation rather than further ‘splitting’.
Darier’s disease (OMIM 124200) is a rare, autosomal dominant genodermatosis in which greasy, yellow to brown, crusted papular lesions develop, mainly in the seborrheic areas of the head, neck, and trunk.1025.1026.1027. and 1028. A rare acral variant exists;1029. and 1030. lesions are sometimes hemorrhagic in this form. 1031 The coalescence of papules produces plaques, which may at times become papillomatous. The prevalence of Darier’s disease varies from 1 : 30 000 in parts of Scotland to 1 : 100 000 in Denmark.1032. and 1033. It is much less common in Singapore. 1034 Onset is usually in early adolescence and the disease runs a chronic course. Congenital onset is rare. 1035 There is an equal sex incidence, but its clinical expression is usually milder in females. 1033 Verrucous lesions resembling acrokeratosis verruciformis may be present on the dorsum of the hands and feet in approximately 70% of cases.1027. and 1036.
Punctate keratoses are sometimes found on the palms and soles. 1036 Longitudinal striations are usually present in the nails. 1037 Rare clinical variants include a bullous,1038. and 1039. a comedonal,1040.1041. and 1042. and a hypertrophic (cornifying) form.1043. and 1044. Comedomal Darier’s has clinical features similar to acne and histopathological features akin to warty dyskeratoma. It is exceedingly rare. 1045 A ‘groversid’ variant with smooth papules has been reported. It was of late onset. 1046 Cutaneous depigmentation (guttate leukoderma),1047.1048.1049.1050. and 1051. secondary eczematization, 1052 cutis verticis gyrata, 1053 mucosal lesions,1054. and 1055. ocular disorders, 1056 bone cysts, 1057 gynecomastia, 1058 and mental deficiency1059 have also been recorded. Mucosal lesions usually involve the mouth but the esophagus has also been involved. 1060 Darier’s disease has been exacerbated and initiated by lithium carbonate therapy1061 and by UVB radiation.1034.1062. and 1063. A variant restricted to sun-exposed areas has been reported. 1064 A paraneoplastic variant has also been reported. 1065 Localized (segmental) disease has been precipitated by the administration of menotropin, 1066 and it has been associated with Gardner’s syndrome in one case. 1067 Patients with segmental, unilateral, or localized lesions are regarded as examples of mosaicism. They were discussed in the introduction to this section (see p. 266). The involved skin has the same genetic mutation that occurs in the generalized form of Darier’s disease. 985 In one case unilateral Darier’s disease involving the entire right side of the body was present. It was associated with unilateral guttate leukoderma. Lesions did not follow Blaschko’s lines as is usual in localized and segmental disease. 1068 The case reported as unilateral Grover’s disease along Blaschko’s lines was probably linear Darier’s disease.1069. and 1070.
There is a predisposition to bacterial, fungal, and viral infections, although no consistent and specific immunological abnormality has been demonstrated.1025.1034.1071. and 1072. Infection with herpes simplex virus, vaccinia, 1073 and even coxsackievirus A161074 may produce the features of Kaposi’s varicelliform eruption1075.1076.1077. and 1078. (see Ch. 26, p. 616). Herpes zoster has occurred in linear Darier’s disease in a patient infected with HIV. 1079 An HPV-related subungual squamous cell carcinoma has been reported. 1080
The mechanism of acantholysis in Darier’s disease is still the subject of controversy. 1025 It is usually ascribed to a defect in the synthesis, organization, or maturation of the tonofilament–desmosome complexes. 1081 Ca2+-ATPases play a key role in the assembly of desmosomes. 1082 It appears that the proteins of the desmosomal attachment plaque are primarily affected.1083. and 1084. There is a loss of desmoplakin I and II and plakoglobin from the desmosomes. 1083 This results from an abnormality in the sorting of desmoplakin. 1082 The pathogenetic significance of the finding that there is a delay in the expression of the suprabasal skin-specific keratins is uncertain. 1085 Loss of desmosomal adhesion triggers anoikis, 1086 a type of apoptosis characterized by cell detachment. This process contributes to the formation of dyskeratotic cells. 1082 The loss of Bcl-2 and Bcl-x in lesional skin of Darier’s disease reflects an imbalance in the apoptotic pathway in this disease.1087. and 1088. The adherens junction is another site of abnormality. 1089
The genetic defect in Darier’s disease has been mapped to chromosome 12q23–q24.1.1090.1091.1092. and 1093. It is due to mutations in the ATP2A2 gene, which encodes the sarco/endoplasmic reticulum Ca2+-ATPase type 2 isoform (SERCA2), a keratinocyte Ca2+ pump.1094. and 1095. Several isoforms of SERCA exist: SERCA2a, SERCA2b, and SERCA3. 1096 Their differential expression may explain the localization of lesions. Variable clinical severity may occur in different members of the same family1097 but no involvement of other organs in which SERCA2 plays a role has been found. 1098 No particular mutational ‘hotspots’ have been found to explain the phenotypic variation that occurs in some pedigrees with Darier’s disease. 1095 Numerous mutations have now been described in this gene.1099.1100. and 1101. In one novel missense mutation, the disease was restricted to the extremities. 1102
Oral retinoids are the most effective treatment for generalized or severe Darier’s disease. The response to topical retinoids is often poor. 1034 Oral retinoids are not without their side effects and some patients prefer to experience the disease itself without the side effects of treatment. 1103 Topical 5-fluorouracil is a promising treatment option for this disease although its use has been limited. 1104 Topical tacrolimus gave encouraging results in one patient forced to cease isotretinoin due to major depression. 1103 Potassium permanganate compresses and topical corticosteroids have been used for inflamed and macerated lesions. 1034 Steroids often control any itch.
An individual papule of Darier’s disease shows suprabasal acantholysis with the formation of a small cleft (lacuna). Irregular projections of the papillary dermis covered by a single layer of basal cells, the so-called ‘villi’, extend into the lacunae (Fig. 9.15). 1025 A thick orthokeratotic plug, often showing focal parakeratosis, overlies each lesion. Mild papillomatosis is often present.
Darier’s disease. There is suprabasal clefting with a thickened stratum corneum and some dyskeratotic cells in the epidermis. (H & E)
Two characteristic types of dyskeratotic cell are present – corps ronds and grains. The corps ronds are found as solitary cells or sometimes small groups of separated cells in the upper malpighian layer and stratum corneum. 1105 They have a small pyknotic nucleus, a clear perinuclear halo, and brightly eosinophilic cytoplasm. The grains are small cells with elongated nuclei and scant cytoplasm in the upper layers of the epidermis. They resemble parakeratotic cells but are somewhat larger (Fig. 9.16).
Darier’s disease. Dyskeratotic cells (corps ronds) are present above the suprabasal cleft. (H & E)
As a general rule, the keratin plug is much thicker in lesions of Darier’s disease, than it is in Grover’s disease. Furthermore, a few eosinophils are almost invariable in the upper dermis in Grover’s disease. They are less common in uncomplicated Darier’s disease.
The keratotic papules on the dorsum of the hands resemble those seen in acrokeratosis verruciformis, but small foci of suprabasal acantholytic dyskeratosis may be seen if serial sections are examined.
The cases reported as comedonal Darier’s disease have had multiple lesions resembling warty dyskeratoma (Fig. 9.17).1040. and 1041.
(A) Comedonal Darier’s disease. (B) There are basal layer downgrowths with villi, resembling those seen in warty dyskeratoma. (H & E)
Immunoglobulins and C3 have been found in the intercellular areas of affected skin by direct immunofluorescence; 1106 this finding has not been confirmed in other studies. 1107 There is premature expression of involucrin in the lower epidermal layers. 1108 There is strong labeling for involucrin in both keratinocyte cell membrane and cytoplasm. 1108
Confocal reflectance microscopy has been used to make the diagnosis. 1109
The corps ronds have a vacuolated perinuclear halo surrounded by a ring of tonofilaments aggregated with keratohyaline granules.1110.1111. and 1112. The grains show premature aggregation of tonofilaments. 1112 The synthesis of keratohyalin in association with clumped tonofilaments is peculiar to Darier’s disease. 1112 It is not seen in Hailey–Hailey disease. As already mentioned, there is controversy over whether the withdrawal of the tonofilaments from the attachment plate of the desmosomes is the primary event in the acantholytic process or merely secondary to the splitting of the desmosomes. 1113 Another ultrastructural finding in Darier’s disease is the presence of cytoplasmic processes projecting from the basal keratinocytes into the underlying dermis through small defects in the basal lamina. 1114
Galli–Galli disease is an acantholytic variant of Dowling–Degos disease, an autosomal dominant disorder characterized by progressive pigmented lesions involving large body folds and flexural areas. 1115 It is due to a mutation of the keratin 5 (KRT5) gene on chromosome 12q13. Mutations in this gene are also responsible for epidermolysis bullosa simplex with palmoplantar keratoderma. 1116 The term ‘Galli–Galli’ is derived from the family name of the two brothers originally reported with this variant. 1115
There are multiple foci of acantholysis within the suprabasal and upper spinous layers with overlying parakeratosis. Dyskeratotic cells are present. Elongate, finger-like strands of keratinocytes extend into the papillary dermis. 1115
The eponymous designation Grover’s disease is the preferred title for several closely related dermatoses characterized by the sudden onset of small, discrete, sometimes crusted, erythematous papules and papulovesicles.1117.1118. and 1119. They usually develop on the upper trunk of older men. A zosteriform variant has been reported. 1120 Lesions may be transient, lasting for weeks or several months1117 (transient acantholytic dermatosis), or they may persist for several years1121 (persistent acantholytic dermatosis1122.1123. and 1124. or papular acantholytic dermatosis1125). Intense pruritus is often present. Oral involvement has been reported, 1126 as has the coexistence of other dermatoses such as asteatotic eczema,1127. and 1128. allergic contact dermatitis, 1128 atopic eczema,1128. and 1129. psoriasis, 1127 pemphigus foliaceus, 1130 a drug reaction, and a neutrophilic dermatosis in association with Waldenström’s macroglobulinemia. 1131
Some cases of Grover’s disease have followed or been exacerbated by exposure to ultraviolet light.1123. and 1125. Cases associated with lentiginous sun-induced ‘freckling’ have been reported.1132. and 1133. A few are associated with chronic renal failure.1134. and 1135. One case involved a febrile postoperative patient. 1136 The role of heat, persistent fever, and sweating has been postulated in the etiology of this condition.1137.1138.1139. and 1140. This theory has been challenged as a consequence of the study performed at the Ackerman Academy of Dermatopathology in New York. 1141 Grover’s disease was present in 0.09% of biopsy specimens. It was diagnosed approximately four times more commonly in winter than in summer. It was suggested that Grover’s disease arises against a backdrop of a xerotic epidermis with decreased sweat production, rather than being caused by sweating and heat as previously postulated. 1141 Both mechanisms are probably involved. The cytokine interleukin-4, the synthetic guanosine analogue ribavirin, 1142 and d-penicillamine therapy1143 have all precipitated Grover’s disease. 1144 Other chemotherapeutic agents, often used in ‘cocktail’ form, have also resulted in Grover’s disease. 1145 Other drugs appear to precipitate this condition, and the author is currently supervising a study of over 300 cases of Grover’s disease to elucidate its clinical associations. In cases thought to have been drug-related, cessation of the drug has not necessarily led to the cure of the Grover’s disease.
Several mechanisms are theoretically possible as explanations for the occurrence of Grover’s disease in cancer patients.1146.1147. and 1148. Several cases appear to have followed induction therapy, and one followed the use of 2-chlorodeoxyadenosine in the treatment of hairy cell leukemia. 1149 Other cases have been in patients who underwent high-dose chemotherapy followed by bone marrow transplantation or autologous stem cell infusion. 1150
Despite the histological similarity to Darier’s disease, Grover’s disease does not share an abnormality in the ATP2A2 gene. 1151 Its pathogenesis is unknown. Loss of syndecan-1 expression occurs, but this is seen in other bullous diseases. Syndecan-1 is a heparan sulfate proteoglycan present on the keratinocyte membrane; it functions in intercellular adhesion. 1152
No single agent, local or systemic, is consistently effective in the treatment of Grover’s disease. 1153 Treatment ranges from emollients, topical corticosteroids, and topical vitamin D analogues, such as calcipotriol and tacalcitol, 1154 to systemic therapies such as oral corticosteroids, retinoids, and PUVA. 1153 In a patient with follicular lymphoma, the Grover’s disease remitted after treatment of the lymphoma with the anti-CD20 antibody rituximab. 1155
Four histological patterns may be seen1127– Darier-like, Hailey–Hailey-like, pemphigus vulgaris-like, and spongiotic (Fig. 9.18 and Fig. 9.19). The Darier pattern was the most common in one series. 1121 In a recent review of 72 cases from the Mayo Clinic, however, the acantholysis resembled pemphigus vulgaris in 40, Darier’s disease in 16, pemphigus foliaceus in 2, Hailey–Hailey disease in 2, and it was spongiotic in 12. 1156 The pemphigus vulgaris type was the most common histological type in the series from the Ackerman Academy of Dermatopathology. 1141 In the spongiotic type there are a few acantholytic cells within and contiguous with spongiotic foci.1122. and 1127. More than one of these histological patterns may be present. In some reports, the persistent cases have tended to have either a Darier-like1123 or pemphigus pattern. 1125 In addition to the epidermal changes, there is usually a superficial dermal infiltrate of lymphocytes and sometimes eosinophils.1121. and 1122. The presence of eosinophils in Grover’s disease serves as a distinguishing feature from Darier’s disease, in which they are usually absent.
Grover’s disease. A few acantholytic cells are present within the suprabasal cleft. There is a mild inflammatory infiltrate in the dermis. (H & E)
Grover’s disease. There is prominent acantholysis resembling Hailey–Hailey disease. (H & E)
Older lesions may have considerable acanthosis and only subtle clefting and acantholysis (Fig. 9.20). They may be misdiagnosed as a solar keratosis or non-specific lesion. Small, non-pigmented seborrheic keratoses seem to be increased in number1157 and are sometimes biopsied instead of the lesions of Grover’s disease. The transient, vesiculobullous variant with a pemphigus foliaceus pattern on histology is best regarded as a variant of pemphigus foliaceus and not of Grover’s disease. 1158 Another histological variant, with acantholysis localized to the acrosyringium, has been reported. 1021 In a patient with Grover’s disease and leukemia cutis, syringoma-like structures were present in the dermis. 1148
Grover’s disease. (A) This lesion was of some duration and accordingly there is conspicuous acanthosis. (B) Clefting is visible in the upper layers of the epidermis in another case. Biopsies of lesions such as this are often misdiagnosed. (H & E)
Direct immunofluorescence is usually negative, 1137 although there are several reports describing variable patterns of immunoglobulin and complement deposition.1122.1159. and 1160.
Ultrastructural changes reflect the light microscopic features with variable degrees of acantholysis, dyskeratosis, and cytoplasmic vacuolization.1161. and 1162. The dyskeratosis is represented by an increase in tonofilaments with some clumping. 1162 There is some loss of desmosomes in the affected area, but the hemidesmosomes of the basal layer are preserved. 1157
Hailey–Hailey disease (familial benign chronic pemphigus – OMIM 169600) is an uncommon genodermatosis with recurrent, erythematous, vesicular plaques, which progress to small flaccid bullae with subsequent rupture and crusting.1163. and 1164. The plaques are well demarcated and spread peripherally, often with a circinate border. Rare clinical forms include papular, 1165 verrucous,1166. and 1167. annular, and vesiculopustular variants. 1168 Targetoid lesions resembling erythema multiforme have been reported. 1169 Nikolsky’s sign may be positive. There is a predilection for the neck, axillae, and intertriginous areas such as the genitocrural, perianal, and inframammary region. Occasionally large areas of the skin are involved. 1170 There are rare reports of involvement of oral, ocular, esophageal, 1171 and vaginal1172 mucous membranes or of lesions limited to the vulva996.1173. and 1174. or perianal1175 region. These latter cases are best included with the cases that are now classified as ‘acantholytic dermatosis of the genitocrural region’ (see p. 266).
Another condition with the histological features of Hailey–Hailey disease and unique clinical features is relapsing linear acantholytic dermatosis, in which there are lesions, following the lines of Blaschko, that wax and wane in a systematic pattern. 1176 Segmental Hailey–Hailey disease limited to the left inframammary area has been reported, an example of type I segmental disease. 1177
Hailey–Hailey disease is inherited as an autosomal dominant condition with incomplete gene penetrance. The responsible gene, ATP2C1, has been mapped to chromosome 3q21–q24. 1178 It encodes a novel Ca2+ pump protein hSPCA1, localized to the Golgi apparatus.1179. and 1180. It has been shown that extracellular calcium homeostasis and functioning Ca2+ pumps play a critical role in regulating differentiation and cell-to-cell adhesion of keratinocytes. 1181 Numerous different mutations have been described.1182.1183.1184.1185.1186. and 1187. Phenotypic variations do not appear to correlate with specific mutations.1188. and 1189. Nearly one-third of cases are sporadic. Onset is usually in the late teens and there is a tendency for the disease to improve in late adulthood. 1190
The chronic course is punctuated by periods of spontaneous remission with subsequent exacerbations. Premenstrual exacerbations have been reported. 1191 Lesions may be induced in genetically predisposed tissues by trauma,1192. and 1193. patch testing, 1194 heat, ultraviolet light, 1195 perspiration and infection with scabies, 1196 bacteria, 1197 herpes virus, 1198 or yeasts. HPV-6 was present in the verrucoid perineal lesions in one patient. 1199 HPV-5 was present in a similar case. 1200 Lesions are often mildly pruritic or burning. They are also malodorous.
Rare associations have included psoriasis,1192.1201. and 1202. Darier’s disease,1203. and 1204. localized bullous pemphigoid, 1205 syringomas of the vulva, 1206 and squamous and basal cell carcinomas.1207.1208. and 1209.
The actual mechanism of the acantholysis in Hailey–Hailey disease appears to be localized in the adherens junction region.1089. and 1210. Initially, the defect was thought to be related to a deficiency in one of the intracellular desmosomal proteins, but recent work has shown that they are all biochemically intact. 1211 They may be dysfunctional in other ways. However, there is dissociation of intra- and extracellular domains of desmosomal cadherin and E-cadherin (an adherens junction-associated protein). 1089 This occurs in both Hailey–Hailey and Darier’s disease, but not in pemphigus. 1089 There is normal expression of the gap junction proteins connexins 26 and 43 in non-lesional skin. 1212 Haploinsufficiency is another mechanism used to explain the pathogenesis of this condition. 1213 Expression of ATP2C1 mRNA is experimentally suppressed following UVB irradiation. 1213 This may be relevant to the clinical presentation of this disease following sunlight exposure. 1213 During acantholysis there is internalization of gap junctions, but this appears to be a secondary process. 1212 In contrast to Darier’s disease, in which abnormal cell adhesion is only demonstrable in clinically involved skin, Hailey–Hailey disease is characterized by a widespread subclinical abnormality in keratinocyte adhesion.1210. and 1214. Cultured keratinocytes from patients with Hailey–Hailey disease and Darier’s disease show altered calcium metabolism. 1215
Dermabrasion and laser therapy have been used with success for localized disease.1216.1217. and 1218. For generalized disease, a variety of systemic and topical agents including corticosteroids and antibiotics have been used. Antifungal agents are used in some cases. 1219 In recalcitrant cases oral retinoids, 1197 cyclosporine or a combination of these two agents, 1220 dapsone, methotrexate, thalidomide, or PUVA therapy have been variably effective. One recalcitrant case was improved by photodynamic therapy with 5-aminolevulinic acid, 1219 and another with alefacept (not approved by the FDA for this purpose, at the time of writing). 1181
Selected cases that have been refractory to other forms of treatment have benefited by excision of affected skin and split-skin grafting using uninvolved skin. 1221 Conversely, if suspensions of keratinocytes from affected skin are placed onto healthy heterologous dermis, devoid of its epidermis, the morphological features of the disease are reproduced in vitro. 1222
In early lesions, there are lacunae formed by suprabasilar clefting, with acantholytic cells either singly or in clumps lining the clefts and lying free within them. The lacunae progress to broad, acantholytic vesicles and bullae (Fig. 9.21). Intercellular edema leading to partial acantholysis gives rise to areas with a characteristic ‘dilapidated brick wall’ appearance (Fig. 9.22). 1223
Hailey-Hailey disease. There is suprabasal clefting with pronounced acantholysis. (H & E)
Hailey-Hailey disease with many acantholytic keratinocytes. (H & E)
Epidermal hyperplasia is commonly seen and this is formed, in part, by downward elongation of the rete ridges. Elongated papillae covered by one or several layers of keratinocytes (‘villi’) may protrude up into the bullae.
Some acantholytic cells are dyskeratotic but they have a well-defined nucleus and preserved cytoplasm in contrast to the degenerating dyskeratotic cells of pemphigus. Corps ronds are infrequent and grains are rare.
Neutrophils are sometimes numerous within the vesicles or in the surface parakeratotic crust. Bacteria may also be present in the crust. 1163 The dermis shows a variable, superficial chronic inflammatory cell infiltrate.
The Hailey–Hailey variant of Grover’s disease has only a narrow vesicle involving no more than a few rete ridges, in contrast to the broad lesions of Hailey–Hailey disease. 1223 Although pemphigus vulgaris usually has less acantholysis and some of its cells show more pronounced dyskeratosis, it can be difficult sometimes to distinguish between it and Hailey–Hailey disease without recourse to immunofluorescence.
Although earlier ultrastructural studies reported that detachment of tonofilaments from desmosomes with the subsequent disruption and disappearance of the latter was the primary event leading to acantholysis, 1224 subsequent studies have shown that the initial event is a series of changes in the microvilli leading to loss of cellular adhesions.1225. and 1226. Desmosomes are then separated and invaginated into cells. Thickened bundles of tonofilaments, sometimes in whorls, are found in cells of the prickle cell and granular layers. 1225
Warty dyskeratomas (follicular dyskeratomas) are rare, usually solitary, papules or nodules with an umbilicated or pore-like center.1227.1228. and 1229. They have a predilection for the head and neck of middle-aged and elderly individuals.1230. and 1231. A subungual and an oral lesion have also been reported.1232.1233. and 1234. Warty dyskeratomas average 5 mm in diameter, although an unusually large example, 3 cm in diameter, has been described. 1235 They occasionally bleed or intermittently discharge cheesy material. 1228 There is no evidence of associated Darier’s or Grover’s disease. 1229 PCR analysis for HPV-DNA is negative. 1229
The lesions in comedonal Darier’s disease have the histological features of warty dyskeratoma.1040. and 1041.
There is a circumscribed, cup-shaped, invaginating lesion extending into the underlying dermis (Fig. 9.23). The central depression is filled with a plug of keratinous material containing some grains. These keratin plugs have sometimes been dislodged, particularly in oral lesions. 1233 The epidermal component shows suprabasilar clefting with numerous acantholytic and dyskeratotic cells within the lacuna. Protruding into the lacuna are villi, which are dermal papillae covered by a layer of basal cells. The papillae contain dilated vessels, occasional melanophages, and a few inflammatory cells; inflammatory cells are also present in the underlying dermis. Pilosebaceous follicles may open into the lesion.
Warty dyskeratoma. A cup-shaped invagination is filled with a keratinous plug which in turn overlies an area of suprabasal clefting. (H & E)
In a study of 46 warty dyskeratomas accessioned in Graz, Austria, three patterns were discerned on scanning magnification. 1229 The majority were cup-shaped, with a small number of cystic and nodular cases. Mixed patterns were also seen. The focal contiguity of many of the lesions to pilosebaceous units, and the presence of differentiation towards the follicular infundibulum led the authors to propose the alternative term of ‘follicular dyskeratoma’ for these lesions. 1229
Corps ronds and grains are better developed in the skin lesions than in those in the mouth. 1233
The plaque-like lesion reported as ‘acantholytic dyskeratotic acanthoma’ had features of both warty dyskeratoma and acantholytic acanthoma. 1236
Hypergranulotic dyscornification is a newly recognized pattern of epidermal dysmaturation. It is analogous to other epithelial patterns of disordered keratinization such as epidermolytic hyperkeratosis and acantholytic dyskeratosis. 1237 To date, the lesions reported have been solitary, scaly papules or nodules on the extremity or trunk.
The nature of this process is uncertain. The following possibilities were considered in the original article – a variant of epidermolytic hyperkeratosis, a variant of maturation in a verruca vulgaris, or a variant of the epidermolytic-like changes seen in ichthyosis hystrix of Curth–Macklin and related disorders (see p. 252).1237. and 1238. It would not be surprising to find incidental, segmental, and generalized variants of this abnormality.
The lesions are exoendophytic with digitate epidermal hyperplasia with hypergranulosis. There are some similarities to a verruca vulgaris. Overlying the thickened granular layer, at the tips of the epidermal papillations, are orthokeratotic mounds of large, eosinophilic corneocytes. Keratohyalin granules are retained within these cells. There is often some basket-weave orthokeratin overlying thick and compacted orthokeratin. A pale basophilic substance is present in the higher spinous layer and granular zone. This appears to be in an intercellular location.
There is a variable lymphocytic infiltrate in the upper dermis.
Colloid keratosis is characterized by the presence of homogeneous eosinophilic masses of variable size and number within the upper layers of squamous epithelia. 1239 It has been seen as an incidental finding in neoplastic and non-neoplastic lesions in the skin and respiratory tract, 1240 as well as in pachyonychia congenita and other onychoses. 1239 Reports have appeared mainly in the non-English and dental literature.
It appears to result from a defect in keratinization with the accumulation of cytokeratin precursors or related protein products. Colloid keratosis has no clinical significance and it is a reaction pattern rather than a disease entity.
Homogeneous and rounded pools of eosinophilic material are found in the upper layers of the epidermis. The material is PAS positive and diastase resistant. Ultrastructurally, it is amorphous and devoid of any filaments.
Colloid keratosis must be distinguished from pagetoid dyskeratosis, which is another incidental histological finding (Fig. 9.24). It is characterized by cells with a pyknotic nucleus with a clear halo and a rim of pale cytoplasm.1241. and 1242. Both entities must be distinguishedfrom clear cell papulosis (see p. 675) in which clear cells containing mucin and keratin are present in the epidermis. 1243 It has been suggested that the clear cells might be precursor cells for cutaneous Paget’s disease. 1243
Pagetoid dyskeratosis. Clear cells are scattered through the upper epidermis. (H & E)
There is a group of rare genodermatoses of late onset in which multiple, discrete, keratotic lesions develop as a result of abnormal keratinization. This group includes hyperkeratosis lenticularis perstans, Kyrle’s disease, multiple minute digitate keratoses, and waxy keratoses. Discrete keratotic lesions associated with palmar-plantar involvement or cornoid lamellation are considered elsewhere in this chapter. Certain acquired lesions such as warts, cutaneous horns, callosities, corns, stucco keratoses, solar keratoses, seborrheic keratoses, and lesions produced by tar may present as discrete keratotic lesions. They are not included in this section, which is concerned essentially with keratotic genodermatoses.
Hyperkeratosis lenticularis perstans (Flegel’s disease – OMIM 144150) 1244 is a rare genodermatosis of late onset in which an abnormality in keratinization results in the development of multiple, discrete, 1–5 mm keratoses.1245.1246. and 1247. An autosomal dominant inheritance is sometimes present.1248.1249. and 1250. The lesions are most prominent on the dorsum of the feet and the anterior aspect of the lower legs, but they may also develop on the thighs, upper limbs, and pinnae.1246. and 1251. The keratoses develop in mid to late adult life and persist. Removal of the spiny scale causes slight bleeding. 1252 A unilateral variant has been described. 1253 This may be a manifestation of the postzygotic mosaicism seen in autosomal dominant skin disease.
Several reports, but not all,1251. and 1254. have documented a decrease in or qualitative defects of the membrane-coating granules (lamellar or Odland bodies) in affected areas of epidermis.1255.1256.1257.1258. and 1259. In one study these granules were normal in old lesions but not found in keratinocytes of early lesions. 1260 It has been suggested that these abnormalities are the basis for the defect in keratinization. 1256
No uniformly effective treatment is available. Emollients, topical 5-fluorouracil, retinoids, topical calcipotriol, and PUVA phototherapy have all been used. 1261
There is a discrete zone of compact, deeply eosinophilic hyperkeratosis, with patchy areas of parakeratosis. 1250 There is some acanthosis at the margins of the lesions but the epidermis at the base of the plaque of keratin is thinned with effacement of the rete ridge pattern. 1246 The malpighian layer may eventually be only three cells thick. The granular layer is usually less prominent in this area. There may be some basal vacuolar change1262 and occasional apoptotic cells. 1246 They were prominent in a case reported by Hunter and Donald. 1263 The superficial dermis has a dense band-like infiltrate of lymphocytes, some of which appear activated. 1257 Some of the small lymphocytes have a nucleus with deep infoldings, resembling Sézary cells. 1264 Many of the cells are CD8+. 1259 Capillary proliferation is sometimes present. In old lesions, the epidermal atrophy is no longer present and the inflammatory infiltrate in the upper dermis is absent. 1260
It is tempting to speculate that the inflammatory infiltrate is an immunological reaction directed against emerging clones of abnormal epidermal cells, in much the same way that this occurs in some cases of porokeratosis. 1265
Studies have shown a reduction in keratohyaline granules and some persistence of desmosomal components in the stratum corneum.1254. and 1264. The disparate findings with regard to membrane-coating granules (lamellar bodies) have been referred to above. 1254 Vesicular bodies, lacking an internal lamellate structure, have been described. 1259
The eponymous designation Kyrle’s disease (OMIM 149500) is preferable to the original designation of hyperkeratosis follicularis et parafollicularis in cutem penetrans. 1266 This controversial entity is regarded by some as a late-onset genodermatosis in which abnormal clones of epidermal cells lead to premature keratinization at the expense of epidermal thickness, with the subsequent introduction of keratinous material into the dermis.1267. and 1268. Others regard it as a variant of perforating folliculitis, a view which is supported by the finding of perforating lesions in patients with chronic renal failure on dialysis, with clinical and histological overlap features between Kyrle’s disease, perforating folliculitis, and even reactive perforating collagenosis.1269.1270. and 1271. It has also been suggested that Kyrle’s disease and Flegel’s disease (hyperkeratosis lenticularis perstans – see above) may be different manifestations of the same disease process.1272. and 1273.
Kyrle’s disease, as traditionally described, consists of hyperkeratotic papules 2–8 mm in diameter, containing a central, cone-shaped plug.1266. and 1274. The papules may be follicular or extrafollicular in location and they may coalesce to form a verrucous plaque. 1275 There is a predilection for the lower limbs, but lesions may also occur on the upper limbs and less often on the head and neck.1274. and 1276. Palmar and plantar surfaces are rarely involved.1277. and 1278. Mucosal involvement is exceedingly rare. 1278 A female preponderance has been noted in some series. 1268 Onset is usually in the fourth decade. A family history has been present in a few cases.1268. and 1278. Kyrle’s disease has been associated with chronic renal failure,1279.1280. and 1281. and with diabetes mellitus, 1280 and more rarely with hepatic dysfunction.1274. and 1282.
There is a keratotic plug overlying an invaginated atrophic epidermis. Focal parakeratosis is present in part of the plug; often there is some basophilic cellular debris, which does not stain for elastin. If serial sections are studied, a focus where the epidermal cells are absent and the keratotic plug is in contact with the dermis will often be seen. An inflammatory infiltrate which includes lymphocytes, occasional neutrophils and sometimes a few foreign body giant cells will be present in these areas. Follicular involvement may be present, particularly in those with chronic renal failure where overlap with perforating folliculitis occurs. Eccrine duct involvement was present in one atypical case reported in the literature. 1277
In Flegel’s disease, in contrast, there is massive orthokeratosis and only focal parakeratosis, but no basophilic debris in the keratin layer. 1268 Also, the inflammatory infiltrate is more conspicuous and usually band-like in distribution.
Multiple minute digitate keratosis is a rare non-follicular disorder of keratinization, also known as disseminated spiked hyperkeratosis. It can occur in four different clinical settings: a familial type with autosomal dominant inheritance,1285.1286.1287.1288. and 1289. a sporadic type,1289.1290. and 1291. a paraneoplastic variant, 1292 and a postinflammatory type.1293. and 1294. Hundreds of minute keratotic spikes develop, usually in early adult life. Cases localized to the palms and soles (spiny keratoderma, palmar filiform hyperkeratosis, 1295 music box spine keratoderma) are probably best classified with the palmoplantar keratodermas.1296.1297. and 1298. There is a predilection for the upper part of the trunk and for the proximal parts of the limbs.1299. and 1300.
A closely related entity, minute aggregate keratoses, 1301 has dome-shaped papules and crateriform or annular lesions in addition to the spicular lesions seen in multiple minute digitate keratoses.
Minute keratotic spikes have been reported as an acquired phenomenon following X-irradiation, 1302 in Crohn’s disease, 1303 and following the use of etretinate. 1304 Follicular spicules have been reported in multiple myeloma. They may be composed of proteinaceus material. 1305 Hair casts were present in another case. 1306
A solitary spiked lesion, representing a vertically growing ectopic nail, may occur on a finger tip. 1307
The spicules are composed of densely compacted, thin stacks of orthokeratotic material, often arising from a finely pointed epidermal elevation. The keratinous spicules are 1–3 mm in height (Fig. 9.25). They are not related to hair follicles. There may be mild underlying epidermal hyperplasia. The dermis is normal. The digitate keratoses that develop following irradiation are characterized by parakeratotic plugs and underlying epidermal invaginations.1302. and 1308. Parakeratotic horns were also present in the patient with Crohn’s disease, referred to above.
Spiked hyperkeratosis. The patient had disseminated spicules on her upper arms. (H & E)
The digitate keratoses reported from the scalp as ‘congenital trichoid keratosis’ consisted of a column of corneocytes with shadow cells suggesting matrical differentiation. 1309
Waxy keratoses (kerinokeratosis papulosa1311) are easily detachable, hyperkeratotic papules with a shiny yellowish ‘waxy’ appearance and an onset in childhood. 1312 The distribution is predominantly truncal and the condition may be familial. 1312 A segmental form, representing mosaicism, has been reported.1311. and 1313. The associated presence of confluent hyperkeratosis at other sites is a variable feature.
Larger papules with a mosaic pattern and acral distribution have been reported under the title ‘mosaic acral keratosis’. 1314
Waxy keratoses of childhood are characterized by marked orthokeratotic hyperkeratosis, tenting/papillomatosis of the epidermis, and some acanthosis. There is some resemblance to confluent and reticulated papillomatosis (Gougerot and Carteaud), see page 505; however, waxy keratoses have more prominent hyperkeratosis.
The group of miscellaneous epidermal genodermatoses includes such disparate conditions as acrokeratosis verruciformis, xeroderma pigmentosum, the ectodermal dysplasias, cutaneous and mucosal dyskeratosis, nevoid hyperkeratosis of the areola, and the peeling skin syndrome.
Acrokeratosis verruciformis of Hopf (OMIM 101900) is an autosomal dominant genodermatosis in which multiple papules, resembling plane warts, develop on the dorsum of the hands and fingers and to a lesser extent on the feet, forearms, and legs.1315.1316.1317.1318. and 1319. Sporadic cases also occur. Onset is usually before puberty, but late onset has been recorded.1315.1316.1317. and 1318. It is more common in males. 1318 Other clinical features include punctate hyperkeratoses of the palms, and nail abnormalities. 1320
Lesions identical to those of acrokeratosis verruciformis develop in a significant number of patients with Darier’s disease (see p. 268). Such lesions may precede, follow or develop concurrently with the onset of the more usual lesions of Darier’s disease. 1321 Acrokeratosis verruciformis has been reported in the relatives of individuals with Darier’s disease. 1322 There is considerable controversy as to the nature of the relationship of these two conditions and also about their relationship to the palmar and plantar keratoses which may accompany either disease. One view is that the acral lesions of Darier’s disease and acrokeratosis verruciformis are separate entities. 1318 This is based on the finding of small foci of acantholytic dyskeratosis in some acral keratotic lesions of Darier’s disease if multiple sections are examined. This is also supported by a recent finding that there is genetic heterogeneity in acrokeratosis verruciformis. In one Chinese study no evidence of any linkage to the ATP2A2 gene on chromosome 12q23–q24 was found. 1320 The contrary view is that both diseases result from a single autosomal dominant genetic defect with variable expressivity of the gene.1321.1323. and 1324. This latter theory was supported by the finding in 2003 of a mutation in the ATP2A2 gene in two patients from a British family with acrokeratosis verruciformis, indicating that it is allelic to Darier’s disease. 1325 As already stated (see above), a Chinese study found no such link. 1320
Sections show hyperkeratosis, regular acanthosis, and low papillomatosis, imparting a regular undulating appearance to the surface (Fig. 9.26 and Fig. 9.27). These changes have been likened to ‘church spires’. There is no parakeratosis, no epidermal vacuolation, and no significant dermal inflammatory infiltrate.
Acrokeratosis verruciformis. There is low papillomatosis (‘church spiring’). Multiple lesions were present, but there were no features of Darier’s disease. (H & E)
Acrokeratosis verruciformis in a patient with Darier’s disease. There is compact orthokeratosis and low papillomatosis imparting an undulating appearance to the epidermis. There is a small focus of acantholytic dyskeratosis. (H & E)
Squamous cell carcinomas have been reported in two cases of long standing. 1318
Xeroderma pigmentosum is a rare, autosomal recessive genodermatosis characterized by deficient DNA repair, photophobia, severe solar sensitivity, cutaneous pigmentary changes, xerosis, and the early development of mucocutaneous and ocular cancers, particularly in sun-exposed areas.1326.1327.1328.1329.1330.1331.1332.1333. and 1334. Dermoscopy has been used to monitor skin lesions in this condition. 1335 Neurological abnormalities are present in up to 20%1336 and these are most severe in the De Sanctis–Cacchione syndrome (microcephaly, dwarfism, choreoathetosis, and mental deficiency – OMIM 278800),1326.1328. and 1337. which in most cases is associated with the XP-A variant of xeroderma pigmentosum. The earliest changes in xeroderma pigmentosum usually develop before the age of 2 with a severe sunburn reaction and the development of multiple freckles with variable intensity of melanin pigmentation and interspersed hypopigmented macules. 1338 Pigmentation often develops on the palms and soles and mucous membranes. Later there is dry, scaly skin (xerosis) with poikilodermatous features. Skin tumors, which include solar keratoses, cutaneous horns, keratoacanthomas, squamous and basal cell carcinomas, basosquamous carcinoma, atypical fibroxanthoma, 1339 malignant melanomas, 1340 and angiomas, may develop in late childhood; patients may ultimately die from the consequences of their tumors. 1336 The development of the cutaneous lesions can be retarded by protection from the sun from birth.986. and 1341. Immunological abnormalities have been present in some patients. 1342
Xeroderma pigmentosum involves both sexes and all races with an incidence of 1 : 250 000 and a gene frequency of 1 : 200. 1326 There is a high incidence of consanguinity.1343. and 1344. Heterozygotes could not, until recently, be reliably demonstrated in the laboratory. 1345 They are asymptomatic, although there is one report of an increased incidence of malignant skin tumors in these individuals. 1326 Prenatal diagnosis of xeroderma pigmentosum can be made by an analysis of DNA repair in cells cultured from the amniotic fluid of women at risk. 1346
There is genetic heterogeneity with at least nine different groups recognized by somatic cell fusion studies – so-called ‘complementation groups’.1326. and 1347. Seven of these groups (labeled A to G) have deficient excision repair of ultraviolet radiation-induced DNA damage (particularly nucleotide excision repair), 1348 while in one (the so-called ‘XP variant’) there is defective ability to convert newly synthesized DNA from low to high molecular weight after UV irradiation (postreplication repair).1326.1349.1350. and 1351. These different complementation groups have different clinical correlations, including differing susceptibility to the various cutaneous tumors. 1352 For example, XP-A is the most severe form and some of this group have the De Sanctis–Cacchione syndrome (see above). The severity of the neurological abnormalities in Japanese patients in this group correlates with the sites of nonsense mutations in the XP-A gene. 1353 XP-E is the mildest form, with late onset and a higher residual capacity to repair UV-induced DNA damage in in-vitro studies.1354. and 1355. XP-F,1356.1357. and 1358. XP-G, 1359 and XP-B are extremely rare; the latter group has in some cases been associated with the Cockayne syndrome (short stature, photosensitivity, deafness, mental deficiency, large ears and nose, and sunken eyes).1326.1329.1360.1361.1362.1363.1364.1365. and 1366. The gene responsible for the severe type 2 Cockayne syndrome (OMIM 133540) is the excision repair cross-complementing group 6 (ERCC6) gene, located on chromosome 10q11, but the classic type 1 Cockayne syndrome (OMIM 216400) is due to mutations in ERCC8 on chromosome 5q12. XP-C is the most prevalent form of xeroderma pigmentosum among North Americans and Europeans. 1367 Squamous cell carcinomas are commonly found in group A, basal cell carcinomas in groups C and E and in the variant form, and malignant melanomas in groups C and D and also in the variant form.1326.1368.1369. and 1370. A new disorder with defective DNA repair distinct from xeroderma pigmentosum or Cockayne syndrome, and associated with UV-induced skin cancers, has been reported. 1371
In one study, a skin fibroblast cell strain from a patient with xeroderma pigmentosum was reported to have shown spontaneous morphological transformation to an anchorage-independent form after serial passage. 1372 This presumably has some significance in the development of tumors in vivo; the finding of reduced natural killer cell activity may have a similar significance. 1373
It is now well established that cultured fibroblasts from patients with xeroderma pigmentosum show defective DNA repair following ultraviolet radiation. This abnormality is also present in keratinocytes and melanocytes cultured from affected patients. 1374 Cultured keratinocytes from patients with xeroderma pigmentosum are more sensitive than normal cells to UVB-induced apoptosis. 1348 The DNA nucleotide excision repair pathway involves at least 28 genes, 11 of which have been associated with clinical diseases. 1375 The defect in xeroderma pigmentosum involves an initial phase in the process of excision repair. 1376
All the genes for xeroderma pigmentosa have now been cloned. The gene responsible for xeroderma pigmentosum group A (XP-A) has been cloned and designated the XPA gene. At least 20 mutations of this gene exist, resulting in a different clinical severity for each mutation group.1345.1377.1378. and 1379. The various genes involved in each of the types of xeroderma pigmentosum are listed in Table 9.5.
Absolute sun protection is the key to the management of this condition.
In the initial stages there are no diagnostically specific features. There may be variability in epidermal melanin concentration, telangiectasia of superficial vessels, and a mild perivascular inflammatory cell reaction. With time, the pigmentary changes are more marked, with areas of prominent melanin pigmentation of the basal, malpighian and spinous layers and pigmentary incontinence. Areas of hypopigmentation, sometimes with epidermal atrophy, may be seen. There is eventually prominent solar elastosis and the development of areas of hyperkeratosis. Keratoses and the other tumors already mentioned eventually develop. Tumors of the anterior part of the tongue have also been reported. 1380
Various changes have been noted. They include irregular nuclear morphology, melanosomes with a high degree of polymorphism, and dilated rough endoplasmic reticulum, vacuoles and disrupted desmosomes in basal keratinocytes. 1381 Fibroblast-like cells may show melanophagic activity. 1381 Structures resembling anchoring fibrils and the basal lamina have been noted in the dilated endoplasmic reticulum of these cells. 1382
The ectodermal dysplasias are an expanding but nevertheless rare group of genodermatoses characterized by a diffuse, non-progressive disorder of the epidermis and at least one of its appendages.1383. and 1384. The epidermal component may involve keratinocytes, melanocytes1385 or Langerhans cells or any combination thereof; the ‘appendageal’ component may affect the hair, sebaceous or eccrine glands, the nails or the teeth. 1386 The ectodermal dysplasias comprise approximately 200 clinically distinct syndromes and the limits of this entity are not clearly defined.1387. and 1388. Their incidence in the United States is approximately 1 : 100 000 births. 1389 Abnormalities of non-ectodermal structures may also be present.
The traditional classification of the ectodermal dysplasias into hidrotic and anhidrotic types is not appropriate for the broad range of abnormalities that may occur in this group. They are now classified on the basis of the presence or absence of trichodysplasia, 1390 dental abnormalities, onychodysplasia, and dyshidrosis. Sometimes more than one such abnormality is present such as the odonto-onycho-dermal dysplasia syndrome (OMIM 257980) which also includes dystrophic nails, hyperhidrosis, palmoplantar keratoderma, and atrophic patches on the malar area. 1391 Other examples are cranioectodermal dysplasia (OMIM 218330) with craniofacial and skeletal anomalies including dental anomalies, 1392 and Ellis–van Creveld syndrome (OMIM 225500) due to mutations in the EVC gene on chromosome 4p16.390. and 1393. It combines chondrodystrophy with central nervous system and urinary anomalies. There may also be dystrophic nails, partial adontia, and multiple frenulae. 1393 The Naegeli–Franceschetti–Jadassohn syndrome (OMIM 161000) affects sweat glands, nails, teeth, and skin. It includes reticulate pigmentation of the skin as an important component and is therefore discussed with other disorders of pigmentation (see p. 297). The gene maps to chromosome 17q21 in the region of the type 1 keratin gene cluster. 1394 Another ectodermal dysplasia, this time with pure hair and nail disturbances (OMIM 602032), is due to a defect in the keratin basic hair protein 5 (KRTHB5) gene on 12q13. 1395 A novel locus has also been described on chromosome 17p12–q21.2. 1396 The autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy syndrome (OMIM 240300) is due to mutations in the autoimmune regulator (AIRE) gene. 1397 Alopecia areata and vitiligo may also be present. Most of the syndromes are extremely rare and of little dermatopathological importance. 1398 Six of them merit further discussion.
In anhidrotic (hypohidrotic) ectodermal dysplasia (OMIM 305100), an X-linked recessive disorder also known as the Christ–Siemens–Touraine syndrome, there is anhidrosis or marked hypohidrosis, complete or partial anodontia, hypotrichosis, and a characteristic facies.696.1399.1400.1401. and 1402. Less frequent manifestations include nail dystrophy, genital anomalies, collodion membrane, 1403 neuroblastoma, 1404 absence of mammary glands, palmoplantar keratoderma, 1405 impaired immunity, 1406 and mental retardation.1407. and 1408.
Immune dysfunction is seen in two genetically distinct syndromes. In one (OMIM 300291) there is a mutation in the NEMO gene on Xq28. 1409 In one case it was accompanied by incontinentia pigmenti, the usual manifestation of this mutation involving the NF-κB pathway. 1410 The other (OMIM 164008) involves the NFκ light chain gene enhancer in B cells inhibitor (NFKBIA) gene at 14q13. 390
A prenatal diagnosis of anhidrotic (hypohidrotic) ectodermal dysplasia can be made by an examination of fetal skin, 1411 or by newer PCR-based methods. Female carriers may show reduced sweating and faulty dentition. 1412 Detecting them can be difficult. 1413 The gene responsible (ED1, EDA) is localized at Xq12–q13.1.390.1414.1415.1416.1417.1418. and 1419. It affects a transmembrane protein expressed by keratinocytes, hair follicles, and sweat glands. 1420 Numerous mutations in this gene have already been described. 1419 The gene responsible for the less frequent autosomal recessive form (OMIM 224900) maps to chromosome 2q11–q13. 1420 It involves the ectodysplasin anhidrotic receptor (EDAR) gene.1416.1421. and 1422.
This syndrome (OMIM 604536), described by McGrath et al in 1997, 1423 is an autosomal recessive disorder, characterized by trauma-induced skin fragility and blistering, palmoplantar keratoderma, abnormal hair growth, perioral erosions, nail dystrophy, and often defective sweating (hypohidrosis).556.1424. and 1425. It results from mutations in the PKP1 gene at 1q32, encoding the desmosomal plaque protein plakophilin 1. 1426 This protein is preferentially expressed in the outer root sheath of hair follicles, but no hair shaft abnormalities have been described so far. 556 Preimplantation diagnosis of this syndrome can be made. 1427
The hidrotic, autosomal dominant variant of ectodermal dysplasia (Clouston’s syndrome – OMIM 129500) is characterized by the triad of alopecia, dystrophic nails, and palmoplantar keratoderma.1383.1428.1429.1430. and 1431. Dental abnormalities may also be present but sweating is normal, in contrast to many other ectodermal dysplasias.
Mutations in the GJB6 gene, at 13q11–q12.1, encoding the gap junction protein connexin 30 have been shown to cause this disorder.1432.1433.1434. and 1435. The patient who presented with alopecia, nail dystrophy, palmoplantar hyperkeratosis, keratitis, hearing difficulty, and micrognathia without GJB6 mutations may represent a new type of hidrotic ectodermal dysplasia. 1436
Orofaciodigital syndrome type 1 (OMIM 311200) is an X-linked dominant disorder which is usually lethal in males.1383. and 1437. It is due to a mutation in the CXORF5 gene at Xp22.3–p22.2. There is a marked reduction in sebaceous glands on the scalp or face, dental dysplasia, evanescent facial milia, cleft lip and palate, and malformation of the digits. Mental retardation may be present. Phenotypically related syndromes include digitocutaneous dysplasia (terminal osseous dysplasia with pigmentary defects – OMIM 300244), characterized by bone anomalies, dental anomalies, dysmorphic features, digital fibromas, atrophic plaques and pigmented skin lesions, and X-linked dominant inheritance. 1438 It is due to mutations in a gene on Xq27.3–q28. 1438 Another related syndrome is oculodentodigital dysplasia (OMIM 164200) due to mutations in the GJA1 gene that encodes connexin 43.
This syndrome (OMIM 115150) is characterized by congenital heart defects, cutaneous abnormalities, and distinctive facial features bearing some resemblance to Noonan syndrome (OMIM 163950).1439.1440. and 1441. Mild to severe mental retardation is usually present. A case with hemihidrosis has been reported, indicating that this syndrome, although not usually included with the ectodermal dysplasias, does meet the criteria for this category of diseases. 1442 Nearly 100 cases have now been reported.
The Costello syndrome (OMIM 218040), which also has phenotypic overlap, has a mutation in the HRAS (the Ras Harvey rat sarcoma) gene on 11p15.5. 1443 Mutations in BRAF, MEK1, and MEK2 also appear to be associated with the CFC phenotype. 1444BRAF mutations are more common than MEK1 or MEK2 mutations. MEK mutations are associated with a milder phenotype. 1443 The phenotype associated with KRAS mutations is highly variable and may be suggestive of Noonan syndrome (sometimes called Noonan syndrome type 3 (OMIM 609942) to distinguish it from type 1 (OMIM 163950)), which is due to mutations in the protein tyrosine phosphatase nonreceptor type 11 (PTPN11) gene on 12q24.1. 390 Mutations in SOS-1 gene are also involved in Noonan syndrome. There is a phenotypic continuum between the CFC syndrome and Noonan syndrome. 1443
There are several clinical syndromes characterized by ectodermal dysplasia in association with clefting of the lip and/or palate. Many of them seem to be caused by different mutations in the p63 gene on 3q27.1389.1445.1446.1447. and 1448. They include the EEC syndrome (ectodermal dysplasia, ectodactyly, cleft lip/palate – OMIM 604292 and 129900),1449. and 1450. the Rapp–Hodgkin syndrome1451 (the additional clinical feature is facial hypoplasia – OMIM 129400) and the Hay–Wells or AEC syndrome (the additional clinical feature is ankyloblepharon, but skin fragility1452 is often present as well – OMIM 106260).1448.1453.1454.1455.1456.1457.1458.1459.1460.1461. and 1462. They are allelic. 1463 Another mutation within the DNA-binding domain of p63 produces ADULT (acro-dermato-ungual-lacrimal-tooth) syndrome (OMIM 103285). 1464 Closely related syndromes include Bowen–Armstrong (OMIM 225000) and CHAND (OMIM 214350) syndromes. 1465
The histological features will obviously vary according to which epidermal and appendageal components are involved.
In anhidrotic ectodermal dysplasia the epidermis is thinned. Eccrine glands are absent or rudimentary, although poorly formed intraepidermal eccrine ducts may be present. 1466 Sweat glands are more likely to be absent in scalp skin than in palmar skin. 1467 Apocrine glands may also be hypotrophic. There is a reduction in pilosebaceous follicles although, paradoxically, foci of sebaceous hyperplasia have sometimes been noted on the upper cheeks. 1468 Other reported features include a reduction in seromucous glands in the respiratory tract, 1469 a reduction in epidermal Langerhans cells, and fragmentation of dermal elastic fibers. As eccrine glands do not develop until 20–24 weeks of gestation, whereas hair follicles should be present at this time, it is the absence of pilar units which is used to make the diagnosis on fetal skin biopsies taken at this period of gestation. 1411
In the ectodermal dysplasia/skin fragility syndrome there is a widening of spaces between keratinocytes in the upper spinous layers with dysadhesion and detachment of the upper epidermal layers. There are some suprabasal clefts. Although described as acantholysis, 1470 it is slightly different with the plane of cleavage occurring immediately on the cytoplasmic side of desmosomes in keeping with the intracellular distribution of plakophilin 1. 556 There may be dyskeratotic cells in the keratoderma, and an increase in catagen/telogen hair follicles. 1470
In hidrotic ectodermal dysplasia there is pronounced hyperkeratosis, particularly of the palms and soles, a normal granular layer, and a normal number of sweat glands. The sweat glands are also normal in number in the orofaciodigital syndrome, but sebaceous glands are diminished or absent. 1437 The epidermis may be somewhat atrophic. No specific histological features have been described in the cardiofaciocutaneous syndrome.
In ectodermal dysplasias with clefting the pilosebaceous follicles are reduced in size and small vellus follicles are present. 1454 A scalp dermatitis with variable histological changes may be present.
There has been a report describing a father and son with brownish papules with central keratotic plugs. 1471 Single cell keratinization (dyskeratosis) was present in the epidermis, as well as in the epithelium of the mouth and the conjunctiva. 1471 A similar condition has been called hereditary benign intraepithelial dyskeratosis. There were prominent oral lesions. 1472 In another case, numerous dyskeratotic cells were present in epithelium of the lips, palate, and gums and on the labial surfaces of the genitalia, as an acquired phenomenon: this condition was referred to as acquired dyskeratotic leukoplakia. 1473 Dyskeratotic cells were also a feature of the cases reported as hereditary mucoepithelial dysplasia (HMD). 1474 HMD is characterized by the triad of non-scarring alopecia, well-demarcated erythema of oral mucosa, and psoriasiform perineal rash. 1475 No genetic mutations have been found but many have been excluded. 1475
The rare condition nevoid hyperkeratosis of the nipple and areola is manifest by hyperpigmentation of the areola with accompanying verrucous thickening. 1476 Over 50 cases have now been reported. It may be unilateral or bilateral.1477.1478. and 1479. Similar, but usually milder, changes may be seen in Darier’s disease, pregnancy and, rarely, in men receiving estrogen therapy for prostatic adenocarcinoma. 1480 Its association with acanthosis nigricans is well documented.1481. and 1482. Cases unrelated to estrogen therapy have been reported in males.1483. and 1484. It has also been reported in a patient with incomplete androgen insensitivity syndrome (46XY karyotype) on estrogen replacement therapy. 1485
There are no trials on the treatment of this condition. All reports are anecdotal. Cryotherapy and topical keratolytics have been used but agents such as 40% urea, 12% lactic acid lotion, and topical retinoids have given mixed or poor results. 1486 Several cases have responded well to calcipotriol.1481. and 1487.
There is hyperkeratosis, papillomatosis, and acanthosis with marked elongation of the rete ridges, the latter often having a filiform interconnecting pattern. Keratin-filled spaces and ostia are also present.1480. and 1488. There is a superficial resemblance to a seborrheic keratosis, but with a more delicate, interconnecting acanthosis. 1489 In another case there was a resemblance to nevus comedonicus. 1490
The peeling skin syndrome (OMIM 270300) is a rare autosomal recessive disorder characterized by spontaneous, continual peeling of the skin.1491. and 1492. Variants in which the peeling has been localized to the palm, 1493 face, 1494 or acral regions1495. and 1496. have been reported. The acral variant (OMIM 609796) is caused by a homozygous mutation in the transglutaminase-5 (TGM5) gene. 1497 Azathioprine has been associated with a clinical simulant. Aminoaciduria was present in another case. 1498
Erythrokeratolysis hiemalis (keratolytic winter erythema, Oudtshoorn disease – OMIM 148370), a rare autosomal dominant genodermatosis with seasonal variation, is common in South Africa and related to an abnormality in chromosome 8p22–p23. It is also characterized by foci of erythema and skin peeling. 239 Spontaneous mutations appear to occur. 1499
There is hyperkeratosis, parakeratosis, reduction of the granular layer, and acanthosis. There is separation of the stratum corneum from the underlying granular layer.1491. and 1500.
In erythrokeratolysis hiemalis there is ‘necrobiosis’ of keratinocytes in the malpighian layer, although only hyperkeratosis and acanthosis were present in another case. 1501
Only three conditions will be considered in this section: granular parakeratosis, circumscribed acral hypokeratosis, and white sponge nevus.
Granular parakeratosis is an acquired abnormality of keratinization first described in 1991 as axillary granular parakeratosis.1503.1504.1505. and 1506. It has since been described in other intertriginous areas such as the inguinal region, 1507 inter- and submammary region, 1508 the vulva and perianal region.1504. and 1508. Multiple intertriginous areas may be involved simultaneously. 1509 Rare cases involving the abdomen and the knee have been seen, indicating that the adjectives ‘axillary’ and ‘intertriginous’ are both inappropriate in the title. Most cases occur in adults but children may also be involved.1510.1511.1512. and 1513. There is a female predominance. 1514 The lesions are scaly red to hyperpigmented plaques that are often pruritic. 1515 Sometimes the lesions appear macerated. 1516 Rarely they are papillomatous. 1514 Axillary lesions can resemble acanthosis nigricans. A case of granular parakeratosis of eccrine ostia presented with small pruritic papules on the face and neck. 1517
Granular parakeratosis has been observed as an incidental finding overlying molluscum contagiosum, 1518 and in association with dermatomyositis, 1519 dermatophyte infection, 1520 and cutaneous carcinomas. 1521 It has also developed in a woman with ovarian carcinoma treated with liposomal doxorubicin. 1522 The lesions spontaneously regressed after 4 weeks.
The cases reported by Resnik and colleagues as granular parakeratotic acanthoma were regarded as akin to epidermolytic acanthoma and acantholytic dyskeratotic acanthoma. 1523 If this analogy is accepted then the cases reported are worthy of acceptance and not dismissible as seborrheic keratoses as suggested by Wang et al. 1517
There appears to be a defect in the processing of profilaggrin to filaggrin that results in a failure to degrade keratohyaline granules. 1504 The etiology is unknown. Suggestions that the condition may result from excess use of topical antiperspirants seem difficult to sustain in cases occurring outside the axilla. 1524 Mechanical irritation may also play a role in inducing these skin changes.1525. and 1526. In several childhood cases, the mothers all reported the habit of frequent washing followed by application of many topical products. 1512 Diaper wearing or the treatment of diaper rashes appears to play an important role in the genesis of infantile cases.1513. and 1527.
Granular parakeratosis usually clears spontaneously after months to one year. 1527 It has cleared rapidly following the topical use of tretinoin. 1528 Other treatments used include 40% urea cream, 1514 topical calcipotriene and ammonium lactate, 1529 and a combination of 1% hydrocortisone cream and 1% clotrimazole cream. 1509
There is a thick parakeratotic layer with retention of keratohyaline granules in this region (Fig. 9.28). The stratum granulosum is also preserved. The underlying epidermis may be normal, mildly atrophic or show psoriasiform acanthosis. The process has also involved the follicular infundibulum1530 and the eccrine ostia. 1517 No hair follicles were involved in this latter case. 1517
Granular parakeratosis. This biopsy, from the axilla, shows a thick parakeratotic layer with retention of keratohyaline granules. (H & E)
Originally described in 2002 as circumscribed palmar or plantar hypokeratosis by Pérez and colleagues, 1531 the word ‘acral’ was suggested by Berk et al in 2007 as a shortened title. 1532 It has merit. This entity has a predilection for the thenar and hypothenar aspects of the palm and, much less commonly, the medial side of the sole.1531. and 1533. Lesions present as circumscribed round to oval erythematous areas of depressed skin.1534. and 1535. In most cases lesions are solitary, but more than one lesion is uncommonly present. 1536 There is a predilection for middle-aged and elderly females.
The etiology is unknown. Chronic repetitive trauma may, in some cases, induce the lesions. 1537 HPV-4 has been isolated from one case, 1538 and HPV-6 in another, but this latter patient had previously been treated for warts in the same location. HPV has been specifically excluded in other reports.1539. and 1540. It more likely represents a localized defect in keratinization on acral sites, morphologically expressed in the granular and horny layers.1540. and 1541.
As the lesions are asymptomatic no treatment is necessary. 1542 Cryotherapy, 1543 photodynamic therapy, 1544 and prolonged treatment with topical calcipotriol1541 have been successfully used.
Parenthetically, a case seen by the author 35 years ago was regarded by the late Dr Hermann Pinkus, to whom the lesion was sent in consultation, as a ‘minus nevus’, a term with some merit.
The lesions show a broad zone of marked hypokeratosis contrasting with the adjacent normal volar skin. Many authors have described a ‘sharp step’ between the involved and uninvolved skin but Resnik and DiLeonardo stated that in their cases the junction (interface) was angled in an irregular and frayed fashion. 1545 The zone of hypokeratosis includes compact orthokeratin atop thin zones of parakeratosis of variable thickness. 1545 There is usually hypogranulosis beneath the parakeratosis. In the zone of hypokeratosis, acrosyringeal corneocytes, when present, are compactly orthokeratotic and protrude slightly above the adjacent stratum corneum. 1545 There is mild pallor of underlying keratinocytes and mild acanthosis. Dilated vessels are present in the papillary dermis and also rare inflammatory cells.
The corneocytes show intracytoplasmic splitting but structures for cell attachment remain intact. 1539
Although white sponge nevus (OMIM 193900) predominantly affects non-cornified stratified squamous epithelia, most often the buccal mucosa, it is considered here because pathogenic mutations in two of the keratin genes, KRT4 and KRT13, have been found in this condition. 1546KRT4 maps to 12q13 and KRT13 to 17q21–q22. These two keratins are normally found in the suprabasal keratinocytes of buccal, nasal, and esophageal mucosa, and anogenital epithelia. 1546
It is an autosomal dominant disorder characterized by thickened spongy mucosa with a white opalescent tint. It involves predominantly the buccal mucosa. It usually presents in early childhood.
The affected mucosa shows epithelial thickening, parakeratosis, and extensive vacuolization of the suprabasal keratinocytes. 1546
Introduction
1. Javier, B; Ackerman, AB, Keratinocyte? Dermatopathology: Practical & Conceptual 7 (2001) 175–178.
2. Haake, AR; Polakowska, RR, Cell death by apoptosis in epidermal biology, J Invest Dermatol 101 (1993) 107–112.
3. McGowan, KM; Coulombe, PA, Keratin 17 expression in the hard epithelial context of the hair and nail, and its relevance for the pachyonychia congenita phenotype, J Invest Dermatol 114 (2000) 1101–1107.
4. Smack, DP; Korge, BP; James, WD, Keratin and keratinization, J Am Acad Dermatol 30 (1994) 85–102.
5. Schweizer, J; Bowden, PE; Coulombe, PA; et al., New consensus nomenclature for mammalian keratins, J Cell Biol 174 (2006) 169–174.
6. Waseem, A; Dogan, B; Tidman, N; et al., Keratin 15 expression in stratified epithelia: downregulation in activated keratinocytes, J Invest Dermatol 112 (1999) 362–369.
7. Porter, RM; Corden, LD; Lunny, DP; et al., Keratin K6irs is specific to the inner root sheath of hair follicles in mice and humans, Br J Dermatol 145 (2001) 558–568.
8. Irvine, AD; McLean, WHI, Human keratin diseases: the increasing spectrum of disease and subtlety of the phenotype-genotype correlation, Br J Dermatol 140 (1999) 815–828.
9. Chu, PG; Weiss, LM, Keratin expression in human tissues and neoplasms, Histopathology 40 (2002) 403–439.
10. Mills, CM; Marks, R, Acral epidermolytic hyperkeratosis, Br J Dermatol 128 (1993) 342–347.
11. Smith, FJD; Irvine, AD; Terron-Kwiatkowski, A; et al., Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris, Nat Genet 38 (2006) 337–342.
12. Ishida-Yamamoto, A; Hashimoto, Y; Manabe, M; et al., Distinctive expression of filaggrin and trichohyalin during various pathways of epithelial differentiation, Br J Dermatol 137 (1997) 9–16.
13. Odland, GF; Holbrook, KA, The lamellar granules of epidermis, Curr Probl Dermatol 9 (1981) 29–49.
14. Mischke, D; Korge, BP; Marenholz, I; et al., Genes encoding structural proteins of epidermal cornification and S100 calcium-binding proteins form a gene complex (‘epidermal differentiation complex’) on human chromosome 1q21, J Invest Dermatol 106 (1996) 989–992.
15. Jonak, C; Metze, D; Traupe, H; et al., The expression of the 27-kd heat shock protein in keratinization disorders: an immunohistological study, Human Pathol 36 (2005) 686–693.
16. Williams, ML, Epidermal lipids and scaling diseases of the skin, Semin Dermatol 11 (1992) 169–175.
17. Williams, ML, Ichthyosis: mechanisms of disease, Pediatr Dermatol 9 (1992) 365–368.
18. Johnson, B; Horn, T; Sander, C; et al., Expression of stratum corneum chymotryptic enzyme in ichthyoses and squamoproliferative processes, J Cutan Pathol 30 (2003) 358–362.
19. Ekholm, IE; Brattsand, M; Egelrud, T, Stratum corneum tryptic enzyme in normal epidermis: a missing link in the desquamation process? J Invest Dermatol 114 (2000) 56–63.
20. Sato, J; Denda, M; Nakanishi, J; et al., Cholesterol sulfate inhibits proteases that are involved in desquamation of stratum corneum, J Invest Dermatol 111 (1998) 189–193.
21. Squier, CA, The permeability of keratinized and nonkeratinized oral epithelium to horseradish peroxidase, J Ultrastr Res 43 (1973) 160–177.
22. Norlén, L; Nicander, I; Rozell, BL; et al., Inter- and intra-individual differences in human stratum corneum lipid content related to physical parameters of skin barrier function in vivo, J Invest Dermatol 112 (1999) 72–77.
23. Sheu, H-M; Chao, S-C; Wong, T-W; et al., Human skin surface lipid film: an ultrastructural study and interaction with corneocytes and intercellular lipid lamellae of the stratum corneum, Br J Dermatol 140 (1999) 385–391.
24. Rossi, A; Jang, S-I; Ceci, R; et al., Effect of AP1 transcription factors on the regulation of transcription in normal human epidermal keratinocytes, J Invest Dermatol 110 (1998) 34–40.
25. Eckert, RL; Crish, JF; Banks, EB; Welter, JF, The epidermis: genes on – genes off, J Invest Dermatol 109 (1997) 501–509.
26. Hohl, D, Towards a better classification of erythrokeratodermias, Br J Dermatol 143 (2000) 1133–1137.
Ichthyoses
27. Rand, RE; Baden, HP, The ichthyoses – a review, J Am Acad Dermatol 8 (1983) 285–305.
28. Williams, ML, The ichthyoses – pathogenesis and prenatal diagnosis: a review of recent advances, Pediatr Dermatol 1 (1983) 1–24.
29. Williams, ML; Elias, PM, Genetically transmitted, generalized disorders of cornification. The ichthyoses, Dermatol Clin 5 (1987) 155–178.
30. Kumar, S; Sehgal, VN; Sharma, RC, Common genodermatoses, Int J Dermatol 35 (1996) 685–694.
31. Bale, SJ; Doyle, SZ, The genetics of ichthyosis: a primer for epidemiologists, J Invest Dermatol 102 (1994) 49S–50S.
32. DiGiovanna, JJ, Ichthyosiform dermatoses: So many discoveries, so little progress, J Am Acad Dermatol 51 (2004) S31–S34.
33. Elsayed-Ali, H; Barton, S; Marks, R, Serological studies of desmosomes in ichthyosis vulgaris, Br J Dermatol 126 (1992) 24–28.
34. Williams, ML; Elias, PM, Ichthyosis. Genetic heterogeneity, genodermatoses, and genetic counseling, Arch Dermatol 122 (1986) 529–531.
35. Bernhardt, M; Baden, HP, Report of a family with an unusual expression of recessive ichthyosis. Report of 42 cases, Arch Dermatol 122 (1986) 428–433.
36. Al-Zayir, AA; Al-Amro Al-Alakloby, OM, Clinico-epidemiological features of primary hereditary ichthyoses in the Eastern province of Saudi Arabia, Int J Dermatol 45 (2006) 257–264.
37. Al-Zayir, AA; Al-Amro Al-Alakloby, OM, Primary hereditary ichthyoses in the Eastern Province of Saudi Arabia, Int J Dermatol 43 (2004) 415–419.
38. Paller, AS, Genetic disorders of skin. A decade of progress, Arch Dermatol 139 (2003) 74–77.
39. Katugampola, RP; Finlay, AY, Oral retinoid therapy for disorders of keratinization: single-centre retrospective 25 years’ experience on 23 patients, Br J Dermatol 154 (2006) 267–276.
40. Verfaille, CJ; Vanhoutte, FP; Blanchet-Bardon, C; et al., Oral liarozole vs. acitretin in the treatment of ichthyosis: a phase II/III multicentre, double-blind, randomized, active-controlled study, Br J Dermatol 156 (2007) 965–973.
41. Pons-Guiraud, A, Dry skin in dermatology: a complex pathophysiology, J Eur Acad Dermatol Venereol 21 (Suppl 2) (2007) 1–4.
42. Sybert, VP; Dale, BA; Holbrook, KA, Ichthyosis vulgaris: identification of a defect in synthesis of filaggrin correlated with an absence of keratohyaline granules, J Invest Dermatol 84 (1985) 191–194.
43. Peña Penabad, C; Pérez Arellano, JL; Becker, E; et al., Differential patterns of filaggrin expression in lamellar ichthyosis, Br J Dermatol 139 (1998) 958–964.
44. Presland, RB; Boggess, D; Lewis, SP; et al., Loss of normal profilaggrin and filaggrin in flaky tail (ft/ft) mice: an animal model for the filaggrin-deficient skin disease ichthyosis vulgaris, J Invest Dermatol 115 (2000) 1072–1081.
45. Nomura, T; Akiyama, M; Sandilands, A; et al., Specific filaggrin mutations cause ichthyosis vulgaris and are significantly associated with atopic dermatitis in Japan, J Invest Dermatol 128 (2008) 1436–1441.
46. Chen, H; Ho, JCC; Sandilands, A; et al., Unique and recurrent mutations in the filaggrin gene in Singaporean Chinese patients with ichthyosis vulgaris, J Invest Dermatol 128 (2008) 1669–1675.
47. McGrath, JA, Filaggrin and the great epidermal barrier grief, Australas J Dermatol 49 (2008) 67–74.
48. Sandilands, A; Terron-Kwiatkowski, A; Hull, PR; et al., Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema, Nat Genet 39 (2007) 650–654.
49. Hoffjan, S; Stemmler, S, On the role of the epidermal differentiation complex in ichthyosis vulgaris, atopic dermatitis and psoriasis, Br J Dermatol 157 (2007) 441–449.
50. Liu, P; Yang, Q; Wang, X; et al., Identification of a genetic locus for ichthyosis vulgaris on chromosome 10q22.3-q24.2, J Invest Dermatol 128 (2008) 1418–1422.
51. Okulicz, JF; Schwartz, RA, Hereditary and acquired ichthyosis vulgaris, Int J Dermatol 42 (2003) 95–98.
52. DiLeonardo, M, Ichthyosis vulgaris vs. X-linked ichthyosis vs. lamellar ichthyosis, Dermatopathology: Practical & Conceptual 2 (1997) 132.
53. Okano, M; Kitano, Y; Yoshikawa, K; et al., X-linked ichthyosis and ichthyosis vulgaris: comparison of their clinical features based on biochemical analysis, Br J Dermatol 119 (1988) 777–783.
54. Honour, JW; Goolamali, SK; Taylor, NF, Prenatal diagnosis and variable presentation of recessive X-linked ichthyosis, Br J Dermatol 112 (1985) 423–430.
55. Paige, DG; Emilion, GG; Bouloux, PMG; Harper, JI, A clinical and genetic study of X-linked recessive ichthyosis and contiguous gene defects, Br J Dermatol 131 (1994) 622–629.
56. Cuevas-Covarrubias, SA; González-Huerta, LM, Analysis of the VCX3A, VCX2 and VCX3B genes shows that VCX3A gene deletion is not sufficient to result in mental retardation in X-linked ichthyosis, Br J Dermatol 158 (2008) 483–486.
57. Ingordo, V; D’Andria, G; Gentile, C; et al., X-linked ichthyosis in Southern Italy, J Am Acad Dermatol 49 (2003) 962–963.
58. Okano, M; Tanaka, H, X-linked ichthyosis associated with congenital dislocation of the hip, Int J Dermatol 40 (2001) 340–342.