For more than 2 decades, animal models have been used to clarify the pathogenic mechanisms of human diseases and develop new therapeutics for these diseases. Several therapies for human diseases have become available through trials using animal models. Epidermolysis bullosa (EB) is one of the most severe inherited skin disorders, whose effective treatments have not been fully available. EB is characterized by abnormalities of the proteins that consist of the dermoepidermal junction. EB has been classified into three major subtypes according to the level of skin cleavage: EB simplex, junctional EB, and dystrophic EB. To date, 13 genes have been shown to cause EB phenotype. After the discovery of the causative genes responsible for each EB subtype, many researchers have tried to develop EB animal models by genetically manipulating the corresponding genes.
For more than 2 decades, animal models have been used to clarify the pathogenic mechanisms of human diseases and develop new therapeutics for these diseases. Several therapies for human diseases have become available through trials using animal models.
Epidermolysis bullosa (EB) is one of the most severe inherited skin disorders, whose effective treatments have not been fully available. EB is characterized by abnormalities of the proteins that consist of the dermoepidermal junction (DEJ). EB has been classified into three major subtypes according to the level of skin cleavage: EB simplex (EBS), junctional EB (JEB), and dystrophic EB (DEB). To date, 13 genes have been shown to cause EB phenotype. After the discovery of the causative genes responsible for each EB subtype, many researchers have tried to develop EB animal models by genetically manipulating the corresponding genes.
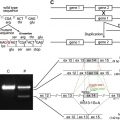
Characterization of animal models of epidermolysis bullosa, and therapeutic experiments using these models
Table 1 summarizes previously described EB animal models whose genetic abnormalities are clarified. The characteristics of each EB animal model and therapeutic experiments using the models are described in this section.
| Disease | Causative Gene | Species | Type | Survival | References |
|---|---|---|---|---|---|
| EBS | KRT5 | Mouse | KO | Neonatal death | |
| EBS | KRT14 | Mouse | Tg | Neonatal death | |
| EBS | KRT14 | Mouse | KO | Neonatal death | |
| EBS | KRT14 | Mouse | KI | Neonatal death | |
| EBS | KRT14 | Mouse | KI (an inducible model) | Not mentioned | |
| EBS-MD/EBS-PA | PLEC1 | Mouse | KO | Neonatal death | |
| EBS-MD/EBS-PA | PLEC1 | Mouse | Conditional KO | Neonatal death | |
| JEB-PA | ITGA6 | Mouse | KO | Neonatal death | |
| JEB-PA | ITGB4 | Mouse | KO | Neonatal death | |
| JEB-PA | ITGB4 | Mouse | KO | Neonatal death | |
| JEB-PA | ITGB4 | Mouse | Partial ablation (expressing ectodomain of β4 integrin) | Neonatal death | |
| JEB-PA | ITGB4 | Mouse | Conditional KO | Not mentioned | |
| nH-JEB | COL17A1 | Mouse | KO | Prolonged survival in 20% of mice | |
| nH-JEB | LAMA3 | Dog | Naturally occurring | Not mentioned | |
| H-JEB | LAMA3 | Mouse | KO | Neonatal death | |
| H-JEB | LAMB3 | Mouse | Naturally occurring | Neonatal death | |
| H-JEB | LAMC2 | Mouse | KO | Neonatal death | |
| H-JEB | LAMC2 | Horse | Naturally occurring | Not mentioned | |
| H-JEB | LAMC2 | Horse | Naturally occurring | Not mentioned | |
| RDEB-sev gen | COL7A1 | Mouse | KO | Neonatal death | |
| RDEB-O | COL7A1 | Dog | Naturally occurring | Not mentioned | |
| RDEB-O | COL7A1 | Mouse | Hypomorphic | Prolonged survival | |
| RDEB-O | COL7A1 | Mouse | KO with transgenic rescue using mutated human cDNA | Prolonged survival | |
| Kindler syndrome | FERMT1 | Mouse | KO | Neonatal death |
Junctional Epidermolysis Bullosa
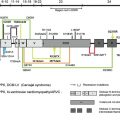
Six genes, COL17A1 , LAMA3, LAMB3, LAMC2, ITGA6, and ITGB4 , have been identified as responsible for the JEB phenotype. Twelve JEB animal models have been developed, although most of the models die perinatally. Recently, the authors’ group developed a Col17a1 knock-out (KO) mice that can survive for approximately 12 months.
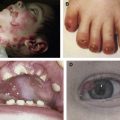
Defective type XVII collagen (COL17) encoded by mutated COL17A1 causes non-Herlitz JEB (nH-JEB). The authors recently developed Col17a1 KO mice whose blistering phenotype is similar to that of human nH-JEB. Notably, 20% of the Col17a1 KO mice exhibited long survival, allowing for the therapeutic experiments, including bone marrow transplantation, cell therapy, protein therapy, and gene therapy. The authors described transgenic rescue experiments for Col17a1 KO mice using human COL17A1 cDNA transgenic mice driven under the keratin-14 promoter, and the rescued mice were born without any skin defects and were able to reacquire reproductive ability. In addition, the tooth abnormalities seen in Col17a1 KO mice were also correctable after incorporation of human COL17A1 cDNA.
Herlitz JEB (H-JEB) is a severe form of EB with short-term survival expectancy. H-JEB is caused by mutations in the genes encoding laminin 332, which consists of the laminin α3, β3, and γ2 chains ( LAMA3/LAMB3/LAMC2 ). Lama3 and Lamc2 KO mouse models have been generated. Furthermore, spontaneous mutant dog, horse, and mouse models whose laminin genes are inactivated have also been described. Those animal models exhibited severe skin detachment with perinatal lethality. Among them, Swiatek and colleagues reported spontaneous mutant mice in which the defective β3 chain of laminin-332 resulted from insertion of an intracisternal-A particle between the exon/intron junction of Lamb3 (Lamb3 IAP mutant).
Schneider and colleagues described prenatal intra-amniotic human LAMB3 cDNA delivery into LamB3 IAP mutant mice using adenovirus and adeno-associated virus vectors. They showed that the defective β3 chain of laminin-332 was expressed in the skin of treated mice, although they found only a minor increase of the lifespan of these mice.
JEB with pyloric atresia (JEB-PA) is caused by mutations in the genes encoding α6 integrin or β4 integrin ( ITGA6/ITGB4 ). α6 or β4 integrin null mice exhibited severe skin detachment with perinatal lethality. Sonnenberg and colleagues generated human ITGB4 cDNA transgenic mice driven under keratin-5 promoter and then tried transgenic rescue for Itgb4 KO mice using those transgenic mice, although the rescued mice still exhibited skin fragility and high mortality.
The same group generated Itgb4 conditional knock-out mice, in which Itgb4 was inactivated only in a small area of the skin. Those mice did not have obvious skin defects, although microscopy showed a small number of blisters in which β4 integrin had been deleted. Giancotti and colleagues generated mice carrying targeted deletion of the β4 integrin cytoplasmic domain. Although those mice expressed truncated extracellular domain β4 integrin protein, their phenotypes were almost the same as mice with complete loss of β4 integrin.
Dystrophic Epidermolysis Bullosa
Mutations in the gene encoding type VII collagen ( COL7A1 ) are responsible for dystrophic EB (DEB). Col7a1 KO mice were developed as a severe generalized recessive DEB (RDEB) model by Uitto and colleagues. Col7a1 KO mice exhibited severe skin detachment with perinatal lethality. Chen and colleagues injected recombinant human type VII collagen (COL7) protein into Col7a1 KO mice and found that the injected human COL7 was incorporated into the DEJ and that the treated mice exhibited longer survival rates than the controls.
Tamai and colleagues reported that embryonic transplantation with bone marrow cells derived from green fluorescent protein (GFP) transgenic mice through vitelline vein–alleviated skin phenotypes of Col7a1 KO mice. Tolar and colleagues also showed that administration of hematopoietic stem-cell–enriched bone marrow cells derived from GFP transgenic mice ameliorated skin fragility and reduced lethality of newborn Col7a1 KO mice. However, the short survival time in which the treatment can show a significant effect in Col7a1 KO mice is still an obstacle to new therapeutics for RDEB.
Bruckner-Tuderman and colleagues developed a DEB hypomorphic mouse model in which the amount of mouse COL7 in skin was approximately 10% of that of wild-type mice and those mice had a prolonged survival. Using this hypomorphic mouse model, they showed that injection of cultured fibroblasts derived from wild-type mice increased the expression of mouse COL7 at the DEJ above the treated area in the hypomorphic mouse skin.
The authors’ group showed transgenic rescue of the previously described Col7a1 KO mouse using two transgenic mouse models comprising human COL7A1 cDNA under keratin-14 promoter or type I collagen promoter. They also used novel methods to develop a milder blistering phenotype in humanized RDEB model mice. They generated human COL7A1 transgenic mice with a premature termination codon (PTC) expressing truncated human COL7 protein. Then, transgenic rescue of Col7a1 KO mice using PTC-causing human COL7A1 transgenic mice resulted in a significantly milder clinical skin blistering manifestation resembling generalized other RDEB and prolonged survival.
Epidermolysis Bullosa Simplex
Mutations in genes encoding keratin 5 and 14 ( KRT5/KRT14 ) have been known to cause EBS, where an abnormal keratin network leads to blister formation. Magin and colleagues developed Krt5 KO mice in which severe skin detachment and perinatal death were noted. Fuchs and colleagues generated transgenic mice expressing a truncated human keratin 14 protein. Those mice exhibited severe skin detachment from the dominant negative effects of aberrantly expressed protein. The same group generated Krt14 KO mice that exhibited a milder phenotype than Krt5 KO mice. Although keratin-null mice showed skin fragility, their mechanism of blister formation is different from that of human patients who have EBS, in which aberrant keratin protein from KRT5/14 missense mutations interferes with the structural assembly of keratin filaments. Roop and colleagues adopted a knock-in strategy to represent the dominant negative effects of abnormal protein encoded by Krt14 harboring a missense mutation in the knock-in mice. They showed that one amino acid substitution is enough to cause an EBS disease phenotype in the knock-in mice. Furthermore, they generated an inducible animal model of EBS, in which topical application of a chemical inducer allows focal activation of a mutant keratin 14 protein in the epidermis of the treated area.
Mutations in the gene encoding plectin ( PLEC1 ) are responsible for EBS with muscular dystrophy and EBS with pyloric atresia. Wiche and colleagues reported that mice with targeted inactivation of Plec1 exhibited severe skin fragility and perinatal death. They also succeeded in obtaining mice with conditional ablation of Plec1 in the stratified epithelia, although those mice also showed markedly short survival.
Kindler Syndrome
The Third International Consensus Meeting on Diagnosis and Classification of EB proposed to include Kindler syndrome within the category of EB. Kindler syndrome is caused by mutations in the gene encoding Kindlin-1 or fermitin family homolog protein 1 (FFH1, FERMT1, KIND1 ). Targeted ablation of Fermt1 failed to show any skin fragility phenotype in mice, although skin specimens from those mice showed skin atrophy.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


