Aging of Skin: Introduction
|
Effects of Aging
In both developed and developing nations, the number and proportion of older people are increasing. In 2008, 23% of the US population was 55 years of age or older.1 The number of persons aged ≥65 years is expected to increase to an estimated 71 million in 2030.1,2 This demographic shift compels health care providers and government officials to confront the pathophysiology of aging and associated health issues.
Aging is a process of progressive decreases in the maximal functioning and reserve capacity of all organs in the body, including the skin. This naturally occurring functional decline in the skin is often compounded and accelerated by chronic environmental insults, such as ultraviolet (UV) and infrared (IR) irradiation as well as environmental carcinogens present in polluted air of major urban centers.
Aging occurs at the cellular level and reflects both a genetic program and cumulative environmentally imposed damage. Mammalian cells can undergo only a limited number of cell divisions and then arrest irreversibly,3–5 in a state known as replicative senescence, after which they are refractory to mitogenic stimuli. This fact has led to the perception that aging evolved in multicellular organisms as a cancer prevention mechanism6 because it prevents the unlimited and possibly unregulated growth of cells whose DNA has been progressively damaged over their life span. Of note, in general the more proficient the DNA repair mechanisms of the organism, the longer its life span.7 Furthermore, there is an inverse correlation between the organism’s life span and metabolic rate,8 consistent with the understood role of cumulative oxidative DNA damage, due to aerobic metabolism, in the aging process.7
Aging Mechanisms
Telomeres, the terminal portions of eukaryotic chromosomes, consist of up to many hundreds of tandem short sequence repeats (TTAGGG in all mammals). During mitosis of somatic cells, DNA polymerase cannot replicate the final base pairs of each chromosome, resulting in progressive shortening with each round of cell division. A special reverse transcriptase, telomerase, can replicate these chromosomal ends, but, with the exception of stem cells and germline cells, the enzyme is normally expressed at extremely low levels. Telomeres of patients with premature aging syndromes, such as Werner syndrome also termed “adult progeria” and associated in addition with increased risk of cancer,10 progeria,11 and dyskeratosis congenita,12 are shorter than those of age-matched controls. Also, most of the DNA repair deficiency diseases, such as xeroderma pigmentosum show varying degrees of “accelerated aging,” or cancer or both.13
Although at low levels, telomerase is expressed in epidermal cells in vivo. In skin, the relatively quiescent fibroblasts and melanocytes have longer telomeres than keratinocytes, but the three cell types exhibit only minor age-dependent telomere shortening of 11–25 bp (base pairs) per year. Some investigators believe that telomerase helps in telomere maintenance of keratinocytes, while dermal fibroblasts and melanocytes maintain their long telomeres due to their low proliferation rate.14,15 Critically short telomeres signal for proliferative senescence or apoptosis, depending on cell type, and appear to compromise DNA stability and transcription of subtelomeric genes,16 presumably contributing to the aged phenotype. Thus, telomeres appear to serve as a biologic clock that determines proliferative life span and functional level of the cell.
In humans, the only genes implicated in the rate of aging are those in which mutations are responsible for premature aging syndromes. For example, Cockayne syndrome patients display mutations in DNA helicases, enzymes that participate in the repair of DNA damage18; ataxia telangiectasia is caused by a mutation in the ATM gene,19 encoding a kinase that detects DNA damage; and Werner syndrome is caused by mutation in a protein with DNA helicase and exonuclease domains.20 Progeria, a disease that leads to death from atherosclerotic heart disease often in the first decade of life, is caused by mutations in lamin A, a protein important in maintaining chromatin organization in the nucleus for transcriptional control and for DNA damage repair.21,22 These human premature aging diseases suggest that decreased DNA repair capacity is associated with accelerated aging and that cumulative DNA damage plays a major role in the aging process. Still, the role of these genes in normal aging is not established, as patients with so-called premature aging syndromes display some manifestations of aging at an accelerated rate but lack other features of normal aging and have characteristic findings that differ greatly from those of normal aging.
Even in DNA repair proficient individuals, throughout life DNA damage accumulates in cells, interfering with cellular metabolism and function. One system that is particularly susceptible to DNA damage is that of growth hormone and insulin growth factor. It is thought that shifting the energy utilization of the cell from growth and proliferation of a damaged cells to preserving the somatic functions of the individual cell has evolved as a way of ensuring the well-being of the organism as a whole and as a cancer preventing mechanism.23 Epigenetic events also play a role in aging. For example, DNA methylation is likely affected with aging and may result in silencing of tumor-suppressor and repair genes resulting in cellular senescence and increasing cancer incidence.24
However, in other species so-called longevity genes have been identified whose mutation or overexpression increase life span. However, in lower organisms all characterized longevity genes encode proteins that assist in control of environmental stress such as starvation, UV irradiation, oxidative damage, and heat shock. Silent information regulator proteins, sirtuins are a class of protein deacetylases implicated in slowing the aging process. Although it is still unclear how they affect aging, it is suggested that they maintain telomere structural integrity, induce transcriptional silencing of genes that promote aging, and/or modulate mitochondrial function in response to caloric restriction.25 Resveratrol, a phenolic substance present in red wine is thought to be sirtuin activator.26 A recently identified transcription factor family of proteins, FoxO, regulates cellular metabolism, stress resistance, and life span extension in mammals.27,28 In addition, FoxO induces the transcription of procollagen I and decreases the transcription of the matrix degrading metalloproteinases (MMP) -1 and -2.28 Mice with extended life spans show high expression of a small number of gene loci that control immune responses, a critical mammalian defense against environmental insults. In the aggregate, these studies strongly support a role of cumulative cellular damage, particularly DNA damage, in the aging process; and proficient repair of such damage in longevity.
The immune system has two major roles: defense against external insults and internal immunologic surveillance. Decreased T cell memory, loss of the naïve T cell population, defective humoral, and cellular immunity characterize the aging immune system. Chronic inflammatory state, decreased immunity to exogenous antigens, and increased autoreactivity compromise the ability to sustain environmental insults.30,31 With aging, increased reactive oxygen species (ROS) within cells leads to oxidative stress and contributes to low-grade inflammation. Additionally, mitochondrial electron transfer during oxidative phosphorylation is compromised with aging and results in leakage of proinflammatory ROS into the cytoplasm. This ROS imbalance contributes to immune senescence beginning with decline in the innate immune response and culminating with impaired adaptive immune responses.31 These changes contribute to the increased incidence of infections and malignancies in the elderly.32
Skin Aging
Cutaneous aging includes two distinct phenomena. Intrinsic aging is a universal, presumptively inevitable change attributable to the passage of time alone; extrinsic aging is the superposition on intrinsic aging of changes attributable to chronic environmental insults, sun exposure, which are neither universal nor inevitable. Extrinsic skin aging is also commonly termed photoaging, reflecting the large and well-studied role of chronic sun exposure. The former is manifested primarily by physiologic alterations with subtle but undoubtedly important consequences for both healthy and diseased skin. The latter has major morphologic as well as physiologic manifestations and corresponds more closely to the popular notion of old skin.
The skin changes that occur with aging (Table 109-1) lead to a gradual physiologic decline (Table 109-2).33 Major age-related changes in the skin’s appearance include dryness (roughness), wrinkling, laxity, and a variety of benign neoplasms. Aged skin is inelastic and recovers more slowly after injury.
Epidermis | Dermis | Appendages |
|---|---|---|
Flattened dermal–epidermal junction | Atrophy (loss of dermal volume) | Depigmented hair Loss of hair |
Variable/decreased thickness | Fewer fibroblasts | Conversion of terminal to vellus hair |
Variable cell size and shape | Fewer mast cells | Abnormal nail plates |
Occasional nuclear atypia | Fewer blood vessels | Fewer glands |
Fewer melanocytes | Shortened capillary loops | |
Fewer Langerhans cells | Abnormal nerve endings |
Barrier function |
Cell replacement |
Chemical clearance |
| DNA repair |
| Epidermal hydration |
| Immune responsiveness |
| Mechanical protection |
| Sebum production |
| Sensory perception |
| Sweat production |
| Thermoregulation |
| Vitamin D production |
| Wound healing |
A major aging theory34 suggests that cumulative damage to biomolecules, including DNA as a result of continuous generation of free radicals, results in increased cellular vulnerability and eventually terminates in senescence or apoptosis. The skin, like other bodily systems, is continuously exposed to ROS generated during aerobic metabolism (eFig. 109-0.1). Although the skin contains a network of antioxidant enzymes (superoxide dismutases, catalase, and glutathione peroxidase) and nonenzymatic antioxidant molecules (vitamin E, coenzyme Q10, ascorbate, and carotenoids), this system is less than completely effective and tends to deteriorate with aging.35
eFigure 109-0.1
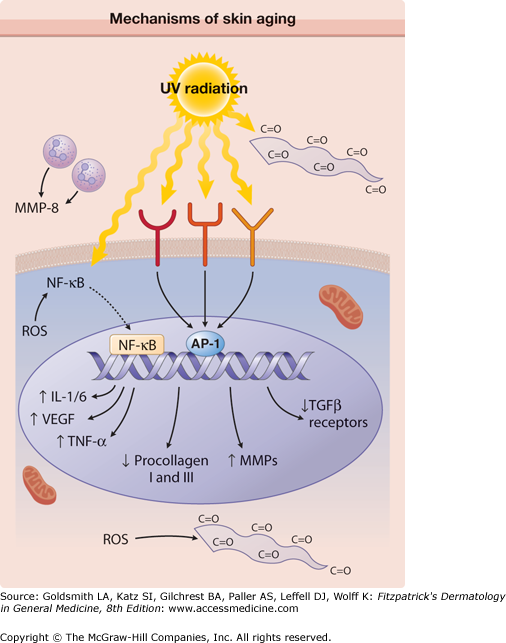
Mechanisms of skin aging. Reactive oxygen species (ROS) generated during aerobic metabolism activate the transcription factor nuclear factor κB (NF-κB) that induces the expression of the proinflammatory cytokines, vascular endothelial growth factor (VEGF), and tumor necrosis factor (TNF)-β. ROS also lead to the formation of carbonyl groups (C = O) in proteins, leading to the accumulation of damaged proteins. Ultraviolet (UV) irradiation directly activates cell surface receptors (indicated by symbols on the cell membrane), initiating intracellular signaling that eventually activates the nuclear transcription complex AP-1. AP-1 increases transcription of matrix metalloproteinases (MMPs) and decreases expression of the procollagen I and III genes and transforming growth factor (TGF)-β receptors, with a final consequence of reduced dermal matrix formation. UV also activates the NF-κB transcription factor that induces the expression of multiple proteins and aggravates the degradation of dermal matrix by increasing MMP levels. Matrix degradation is further exacerbated by MMP-8 (collagenase) of neutrophil origin, following neutrophil infiltration into UV-irradiated skin. Mitochondria display large DNA deletions and compromised function. Damaged proteins containing carbonyl groups accumulate in the upper portions of the dermis. (From Halachmi S, Yaar M, Gilchrest BA: Advances in skin aging/photoaging: Theoretic and practical implications (Part I). Ann Dermatol Venereol 132:362, 2005, with permission.)
Oxidative stress upregulates the level of stress regulatory proteins, including hypoxia-inducible factors (HIFs) and nuclear factor κB (NFκB). HIFs influence the expression of genes that regulate cellular metabolism, survival, motility, basement membrane integrity, angiogenesis, hematopoiesis, and other functions.36 Both HIFs and NFκB induce the expression of proinflammatory cytokines like interleukin (IL)-1 and IL-6, vascular endothelial growth factor (VEGF), and tumor necrosis factor (TNF)-α. These proteins are involved in immunoregulation and cell survival,37 stimulate the expression of matrix-degrading metalloproteins,38 and are believed to play a central role in the aging process. Furthermore, HIFs stabilize subpopulations of malignant cells with stem cell properties (cancer stem cells) and induce their self-renewal by stimulating the expression of signaling pathways critical for survival and proliferation. This suggests that age-associated cellular hypoxia could be involved in cancer stem cell maintenance.36
Oxidative damage also affects telomeres. A recent hypothesis suggests a common cellular signaling pathway activated by DNA damage and involving the terminal portion of the telomeres.39,40 The terminal portion of the 3′ telomeric strand extends beyond the complementary 5′ strand (Fig. 109-1), leaving a single stranded G-rich overhang. It is suggested that during both telomere shortening and repair of telomere damage, such as that encountered during oxidative stress, the normal loop structure at the end of telomeres is disrupted, exposing the 3′ overhang that under baseline conditions is “buried” in the loop structure.39,40 Exposure of the TTAGGG tandem repeat sequence then appears to activate p5341 and to stimulate responses known to include proliferative senescence and apoptosis.40,41 Thus, the intrinsic component of skin aging involves progressive oxidative stress and telomere signaling as telomeres shorten during serial cell division and in response to oxidative DNA damage.39
Figure 109-1
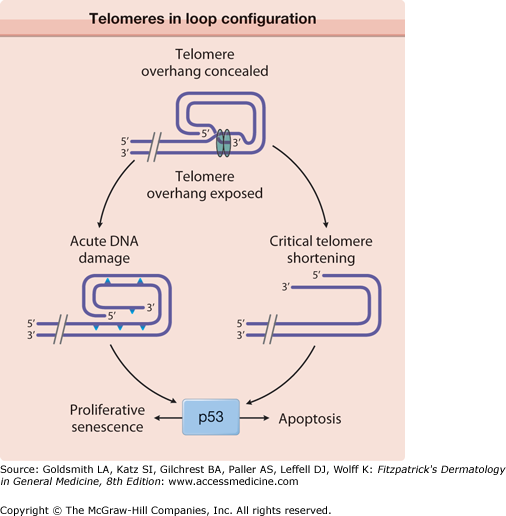
Telomeres normally exist in a loop configuration, held in place by the final 150–200 bases (TTAGGG repeats) on the 3′ strand that forms a single-stranded overhang. When the loop is disrupted when telomeres become critically short (e.g., after repeated cell divisions or when telomeres are damaged as a result of UV irradiation or oxidative damage), the overhang becomes exposed, activating the tumor suppressor protein p53 to induce proliferative senescence or apoptosis, depending on the cell type. IL = interleukin. (From Yaar M: Clinical and histological features of intrinsic versus extrinsic skin aging. In: Skin Aging. Springer, 2006, p. 9, with permission.)
Oxidative damage also affects cellular proteins, leading to the formation of multiple carbonyl groups (C = O). Such proteins are typically targeted for degradation by proteasomes whose function declines with age, leading to the accumulation of damaged proteins that interfere with proper cellular function.42
Another mechanism that plays a role in intrinsic aging is cellular senescence, the limited capacity of cells to divide. It is regarded by some as having evolved in multicellular organisms as a cancer-prevention mechanism.6 Senescent cells display critically short telomeres, irreversible growth arrest, resistance to apoptosis, and altered differentiation. They also overexpress genes that block progression into the cell cycle43–45 as well as genes encoding proteins such as fibronectin and proteases involved in modulation of extracellular matrix, such as collagenase and stromelysin. In addition, the levels of certain tissue inhibitors of MMPs are decreased.46
Additional mechanisms include amino acid racemization, a process that substitutes D-amino acids for L-amino acids within proteins, affecting protein function and rendering them less susceptible to degradation. Finally, nonenzymatic glycosylation of proteins occurs when reducing sugar aldehydes condense with protein amino groups, resulting in brown discoloration, loss of function, and altered degradation. Glycosylation of extracellular matrix proteins, such as dermal collagen, leads to cross-linking with trapping and sequestration of other unaffected proteins.
Many of the morphologic and functional age-associated changes in skin were documented many years ago47 and are not specifically referenced here. The most striking and consistent histologic change is flattening of the dermal–epidermal junction with effacement of both the dermal papillae and epidermal rete pegs.48 This results in a considerably smaller surface between the epidermis and dermis and presumably less communication and nutrient transfer. Dermal–epidermal separation has been demonstrated to occur more readily in old skin, undoubtedly explaining the propensity of the elderly to torn skin and superficial abrasions after minor trauma.
There is an age-associated epidermal thinning of 10%–50% between the ages of 30 years and 80 years.49 Variability in epidermal thickness and individual keratinocyte size increases, including those of the basal layer. Evidence suggests that epidermal keratinocytes senesce and senescent cells are more resistant to apoptosis. Therefore, such keratinocytes are more likely to accumulate mutations, increasing their risk for malignant transformation. Epidermal stem cells are a population of cells responsible for epidermal maintenance. It is unclear whether there is an age-associated decrease in epidermal stem cells. Some studies also show loss of epidermal stem cell population in aged skin as determined by the loss of cells expressing CD71 (transferrin receptor) and α6 integrin, accepted markers for keratinocyte stem cells,50 while others claim that unlike stem cells from other tissues, epidermal stem cells maintain their number and functionality with age and do not display ROS increases.51,52 The latter has been attributed to high levels of antioxidant enzymes particularly superoxide dismutase-1.53 At the electron microscopic level, sun-protected old skin is characterized by some widening of interkeratinocyte spaces, by reduplication of the lamina densa and anchoring fibril complex in the basement membrane zone, and by loss of the numerous microvillous projections of basal cell cytoplasm into the dermis.49
Average thickness and degree of compaction of the stratum corneum appear constant with increasing age, although individual corneocytes become larger. The skin surface pattern, a patchwork of fine lines possibly determined by papillary dermal architecture, reveals slight age-associated loss of regularity. There is an overall decreased lipid content in the stratum corneum of the elderly as well as decreased water content in part as a result of decrements in cholesterol synthesis.54 Age-associated increase in stratum corneum pH impedes lipid-processing enzyme activity.55 Age effects on percutaneous absorption depend in part on drug structure, with hydrophilic substances such as hydrocortisone and benzoic acid being less well absorbed through the skin of old versus young individuals but with hydrophobic substances such as testosterone and estradiol being equally well absorbed.56 Of perhaps greater clinical importance, aging markedly delays the recovery of barrier function in damaged stratum corneum, apparently because of slow replacement of neutral lipids, leading to decreased amount of lipids in the newly formed lamellar bodies.57 Lipid synthesis and activities of enzymes required to generate stratum corneum lipids decrease with age possibly because of aberrations in elements that regulate enzyme transcription, or abnormal autocrine/paracrine signaling.58
In the elderly, the skin often appears dry and flaky, especially over the lower extremities, an area in which a remarkable age-associated decrease in the content of epidermal filaggrin has been reported.59 Filaggrin, required for binding of keratin filaments into macrofibrils, is also decreased in the skin of patients with ichthyosis vulgaris, and its lack has been postulated to cause the increased scaliness in both conditions.59 Barrier function also may be affected by this structural change.
Epidermal turnover rate and thymidine-labeling index decrease approximately 30%–50% between the third and eighth decades, with a corresponding prolongation in stratum corneum replacement rate. Linear growth rates also decrease for hair and nails. Epidermal repair rate after wounding likewise declines with age.
A decrease in the number of enzymatically active melanocytes per unit surface area of the skin, approximately 10%–20% of the remaining cell population each decade, has been documented repeatedly, presumably reducing the body’s protective barrier against UV radiation. Age-associated decline in DNA repair capacity compounds the loss of protective melanin and increases the risk for skin cancer development. The number of melanocytic nevi also decreases progressively with age, from a peak of 15 to 40 in the third and fourth decades to an average of four per person after age 50 years; such nevi are rarely observed in persons beyond age 80.
Between early and late adulthood there is a 20%–50% reduction in the number of morphologically identifiable epidermal Langerhans cells, the skin’s immune effector cells responsible for antigen presentation. The remaining cells display morphologic abnormalities, including less and shorter dendrites, and they display reduced antigen-presenting capacity.60 These changes, compounded by decreases in cytokine production by keratinocytes and lymphocytes and failure of migration through the lymphatic system, presumably contribute to the observed age-associated decrease in cutaneous immune responsiveness.
An endocrine function of human epidermis that declines with age is vitamin D production.61 Vitamin D, by binding its nuclear receptor, induces the transcription of numerous genes.62 Vitamin D deficiency in adults leads to osteomalacia and low levels have been implicated in epidemiologic studies as contributing to diabetes, hypertension, and prevalent tumors.63 Aside from its well-studied role in calcium homeostasis, vitamin D, when bound to its nuclear receptor (1,25D-VDR) influenced transcription of numerous genes including those that encode proteins of the Wnt signaling pathway affecting the formation of the cornified epithelium as well as hair growth. 1,25D-VDR also activates genes that encode proteins that participate in the innate and adaptive immune responses and repress IL-17, a major inducer of autoimmune disorders such as type I diabetes mellitus, multiple sclerosis, lupus, and rheumatoid arthritis. 1,25D-VDR is also anti-inflammatory, as it decreases NFkB and COX2 activation. Finally, 1,25D-VDR induces the activity of the tumor suppressor p53 and p21 proteins and the activity of FoxO, preventing oxidative damage and inducing DNA repair enzymes in skin.64 Elderly individuals frequently have reduced serum levels of vitamin D. Although avoidance of dairy products (the principal dietary source of vitamin D), insufficient sun exposure, and sunscreen use undoubtedly contribute to vitamin D deficiency in the elderly, the level of epidermal 7-dehydrocholesterol per unit skin surface area also appears to decrease linearly by approximately 75% between early and late adulthood,61 suggesting that lack of its immediate biosynthetic precursor also may limit vitamin D production. Together these observations suggest that age-associated decrease in Vitamin D could accelerate the aging process and argue for use of vitamin D dietary supplements in the elderly.65
With regard to susceptibility to oxidative damage, there is progressive accumulation of damaged cellular proteins and lipids with aging.49,66 Furthermore, antioxidant defense systems decline with age, and, in addition, there is a decrease in DNA damage repair capacity.49 These changes in combination increase cellular mutability or their tendency to become senescent, or both.
Loss of dermal thickness approaches 20% in elderly individuals, although in sun-protected sites significant thinning occurs only after the eighth decade.67 Old dermis is relatively acellular and avascular, and there is age-related loss of normal elastic fibers and dermal collagen.68,69
Decreased inflammatory responses in the elderly are the result of decreased synthesis and secretion of keratinocyte-derived cytokines and inflammatory mediators in addition to decreased endothelial response. The dermal microvasculature in middle-aged or elderly subjects also may show mild vascular wall thickening, especially in the lower legs as a result of gravitational forces70; vascular wall thinning to less than one-half the normal young adult measurement, associated with absent or reduced perivascular veil cells, has been reported in skin of very elderly subjects and probably contributes to vascular fragility. Loss of elastin contributes to vascular rigidity. Electron microscopic studies show focal degeneration of the elastic component of dermal arterioles. The striking age-associated loss of vascular bed, especially of the vertical capillary loops that occupy the dermal papillae in young skin, and increased distance from the epidermis of existing loops, is thought to underlie many of the physiologic alterations in old skin, including pallor, decreased skin temperature, and the approximately 60% reductions in basal and peak induced cutaneous blood flow.71
VEGF of epidermal origin appears to play a major role in maintaining dermal vasculature, inducing the expression of antiapoptotic proteins in endothelial cells,70 and decreased VEGF level shown in aged mice and rabbits skin probably contributes to endothelial cells apoptosis.72,73 Also, evidence suggests that there is an age-associated decline of both angiogenic and antiangiogenic factors, disrupting cutaneous angiogenic homeostasis.74 Decreased endothelial cell permeability response and decreased capacity to induce white cell adhesion75 contribute to the compromised immune response. When exposed to intense heat or cold, aging vessels demonstrate reduced ability to constrict, dilate, or shunt.70 Compromised thermoregulation, which predisposes the elderly to sometimes fatal heat stroke or hypothermia, may be due in part to reduced vasoactivity of dermal arterioles and, in the latter instance, to loss of heat-conserving subcutaneous fat as well. Reduction in the vascular network surrounding hair bulbs and eccrine, apocrine, and sebaceous glands may contribute to their gradual atrophy and fibrosis with age.
Age-associated decreases in wheal resorption and dermal clearance of transepidermally absorbed materials have been reported,56 probably due to alterations in both the vascular bed and the extracellular matrix. Conversely, the time required for development of a tense blister after topical ammonium hydroxide application is nearly twice as long in older individuals, suggesting a decreased transudation rate with age in injured skin. Impaired transfer of cells as well as solutes between the extravascular and intravascular dermal compartments is suggested by several studies; multiple factors undoubtedly contribute.
With aging there is a decrease in the density and lumen size of lymphatic vessels accompanied by increased rigidity and decrements in lymphatic drainage, affected by decreased surrounding elastic fibers.70 The ability to effectively pump lymph from interstitial spaces into the lymphatics is impaired with aging in part because of decreased activity of enzymes that catalyze the production of nitric oxide.76
Biochemical changes in collagen, elastin, and dermal ground substance lead to increased skin rigidity primarily due to modifications in collagen. Collagen content per unit area of skin surface decreases approximately 1% per year throughout adult life,77 and the remaining collagen fibrils appear disorganized, more compact, and granular, and they display increased collagen cross-links.78–80 The latter is the result of decreased collagen I and III synthesis; decrements in enzymatic processing of collagen as well as nonenzymatic glycosylation, a process that leads to molecular damage of proteins with a long half-life such as collagen49; and increased collagenase levels. Such changes almost certainly contribute to impaired wound healing in the elderly.79
Beginning in early adulthood, elastic fibers decrease in number and diameter; by old age, they often appear fragmented, with small cysts and lacunae, especially near the dermal–epidermal junction81 most likely due to enzymatic degradation of elastin. Elastic fibers also show progressive cross-linkage and calcification with age. At the biochemical level, there is an age-associated decrease in numerous elastic fiber components, including elastin, fibrillin, and fibulin-2. With aging, the level of fibulin-5, an extracellular matrix protein that functions as a scaffold for elastic fibers, appears to decrease before other changes are observed, suggesting that loss of fibulin-5 is a marker for skin aging.82
The ground substance mucopolysaccharides, glycosaminoglycans (GAGs), and proteoglycans are decreased relative to dry weight or collagen content of the skin, especially hyaluronic acid,83 possibly due to decreased hyaluronan secretion or due to decreased hyaluronic acid extractability.84 Aging also affects GAG composition and binding to elastin, impeding the drainage of molecules into lymphatic vessels.70 These changes may adversely influence skin turgor because proteoglycans bind 1,000 times their own weight in water and also impact collagen fiber deposition, orientation, and size.85
Changes with age in the mechanical properties of the skin during adulthood include progressive loss of elastic recovery, consistent with gradual destruction of the dermal elastic network, and marked prolongation of the time required for excised skin to return to its original thickness. In vivo ultrasound studies also show age-associated differences in water distribution in the dermis,86 no doubt affecting dermal pliability, resilience, and elasticity. Overall, a picture emerges of aging dermis as an increasingly rigid, inelastic, and unresponsive tissue that is less capable of undergoing modifications in response to injury or stress.
Like other striated muscles, facial muscles show accumulation of the “age pigment” lipofuscin, a marker of cellular damage. Compounded by diminished neuromuscular control, this deterioration contributes to wrinkle formation.87 In addition, subcutaneous fat is depleted from distinct facial regions, including the forehead, preorbital, buccal, temporal, and perioral regions. In contrast, there is a prominent increase in fatty tissue in other areas, including the submental regions, the jowls, the nasolabial folds, and the lateral malar areas. In contrast to the young face in which fat is diffusely dispersed, fat in the aged face, subject to the force of gravity, contributes to sagging and drooping of the skin.88
Finally, like other parts of the skeleton, facial bones display reduced mass with age. Bone resorption affects particularly the mandible, maxilla, and frontal bones. Bone loss in these areas enhances the sagging of facial skin and contributes to the obliteration of the demarcation between the contour of the jaw and the neck that is so distinct in young adults.89
By the end of the fifth decade, approximately half the population has at least 50% gray (white) scalp hair, and virtually everyone has some degree of graying due to progressive and eventually total loss of melanocytes from the hair bulb.90 Loss of melanocytes is believed to occur more rapidly in hair than in skin because the cells proliferate and manufacture melanin at maximal rates during the anagen phase of the hair cycle, whereas epidermal melanocytes are comparatively inactive throughout their life span. More specifically, hair graying reflects loss of the melanocyte stem cell population in hair follicle bulge due, at least in part, to compromised interaction between two transcription factors, microphthalmia-associated transcription factor (Mitf) and Pax3 (see Chapter 72).91 Faulty migration of melanocyte stem cells into the bulb area of the hair92 has also been suggested to contribute. Additionally, high levels of H2O2 in the millimolar range have been reported in gray/white scalp.93 By oxidizing methionine, tryptophan, and cysteine residues on enzymes, H2O2 likely to interferes with the activity of tyrosinase as well as antioxidant enzymes by altering their tertiary structure and may thus affect melanogenesis in the human hair follicle.93
Scalp hair may gray more rapidly than other body hair because its anagen to telogen ratio (see Chapter 86) is considerably greater than that of other body hair. Advancing age is also accompanied by a modest decrease in number of hair follicles, due in part to atrophy and fibrosis. In addition, with aging there is an increase in the proportion of telogen hair follicles. Remaining hairs may be smaller in diameter and grow more slowly. One hypothesis suggests that melanocyte loss and lack of melanosomal transfer may increase oxidative stress level in highly metabolic hair follicle keratinocytes, affecting their function and viability.94
The process termed balding results primarily from the androgen-dependent conversion of the relatively dark, thick, terminal scalp hairs to lightly pigmented short, fine, vellus hairs similar to those on the ventral forearm. Women are affected less often and far less severely than men. However, in postmenopausal women, hair loss is also the result of decreased estrogen levels and estrogen to androgen ratio.95,96 Besides hair loss, almost 50% of women older than age 60 years display mild facial hirsutism, presumably attributable to the same hormonal changes as scalp hair loss. In susceptible women, testosterone and/or progestin derivatives that are present in some hormone replacement regimens may exacerbate these changes.
Eccrine glands decrease by approximately 15% in average number during adulthood in most body sites. Spontaneous sweating is further reduced by more than 70% in healthy older subjects compared with younger controls, attributable primarily to a decreased output per gland, predisposing the elderly to heat stroke. Apocrine gland size and function also decrease with aging. Sebaceous gland size and number appear not to change with age, but there is an exponential decrease in sebum production in both men and women most likely due to a decrease in production of gonadal or adrenal androgens.97
Pacinian and Meissner’s corpuscles, the cutaneous end organs responsible for pressure perception and light touch, progressively decrease to approximately one-third their initial average density between the second and ninth decades of life and display greater size variation and structural irregularities.
Decreased sensory perception in old skin encompasses optimal stimulus for light touch, vibratory sensation, and corneal sensation; ability to discriminate two points; and spatial acuity.98,99 Cutaneous pain threshold increases up to 20% with advancing adult age, and compromised arteriolar constriction on changing position from supine to standing is reflective of decreased responsiveness of the sympathetic nervous system.
Estrogens play a critical role in female development and reproduction and also influence skin and hair. Not surprisingly, their influence decreases dramatically after menopause. Menopause typically occurs in a woman’s early 50s, so that, with life expectancy in the developed world approaching 80 years,100 women are postmenopausal for approximately one-third of their lives. In premenopausal women, the predominant estrogen is estradiol, which is produced by the ovaries,101 and after menopause levels decrease by more than 90%,102 with estron, a less active estrogen, becoming the predominant form.103 Progestin and androgen levels also fall markedly after menopause.103 The reduced levels of estrogen underlie many physiologic effects, including hot flashes, atrophy of reproductive tissue, and changes in nonreproductive tissues that are estrogen sensitive.103 Age-associated decrements in keratinocyte barrier function, immune-regulation, and wound healing appear to be compounded by decreased estrogen levels and/or decreased responsiveness of cells to existing estrogens. Because both estrogen and androgen receptors are expressed by skin-derived cells, both hormones are likely to play a role in skin structure and function.
Decreased circulatory levels of estrogens are associated with reduced dermal collagen content,104–106 increased cutaneous extensibility,107,108 and decreased elasticity.109 Also, decreased water-holding capacity, increased dryness, and increased fine wrinkling are reported after menopause,110 as are decreased sebum levels.111 These changes are related more to menopause than to chronologic age alone,112 and wrinkling is reported to be more pronounced in postmenopausal women who are not taking hormone replacement therapy than in treated women.113
After menopause, the decreased rate of wound healing is associated with reduced levels of collagen I.114 Estrogen and progesterone are also reported to modulate cutaneous inflammation, enhance keratinocyte proliferation and collagen synthesis, decrease the activity of MMPs, and increase the synthesis of dermal mucopolysaccharides and hyaluronic acid.96,115
Clinical and histologic features of actinically damaged skin are listed in Table 109-3. A prominent feature of photoaged skin is elastosis, a process characterized clinically by yellow discoloration and a sometimes pebbly surface (Fig. 109-2) and histologically by tangled masses of degraded elastic fibers that further deteriorate to form an amorphous mass composed of disorganized tropoelastin and fibrillin (eFig. 109-2.1A). Although fibrillin is abundant in the elastotic material deeper in the dermis, in the upper portions of the dermis at the dermal–epidermal junction, fibrillin is reduced.116 In addition, the amount of ground substance, largely composed of glycosaminoglycans (GAGs) and proteoglycans, increases in photodamaged skin, whereas the amount of collagen decreases, in part because of increased metalloproteinase activity. In contrast with aged sun-protected skin that demonstrates hypocellularity, photodamaged skin frequently displays an increased number of hyperplastic fibroblasts as well as increased inflammatory cells (eFig. 109-2.1B), including mast cells, histiocytes, and other mononuclear cells, giving rise to the term heliodermatitis (literally, “cutaneous inflammation due to sun”; Fig. 109-3). Immunohistologic studies show increased CD4+ T cells in the dermis. Dermal vasculature in mildly photodamaged skin displays venule wall thickening; in severely photodamaged skin, thin vessel walls with compromised perivascular veil cells display dilations (telangiectases).
Clinical | Histologic |
|---|---|
Dryness (roughness) | Increased compaction of stratum corneum, increased thickness of granular cell layer, reduced epidermal thickness, reduced epidermal mucin content |
Actinic keratoses (see Chapter 113) | Nuclear atypia, loss of orderly, progressive keratinocyte maturation; irregular epidermal hyperplasia and/or hypoplasia; occasional dermal inflammation |
Irregular Pigmentation | |
Freckling | Reduced or increased number of hypertrophic, strongly DOPA-positive melanocytes |
Lentigines (see eFig. 109-2.2) | Elongation of epidermal rete ridges; increase in number and melanization of melanocytes |
Guttate hypomelanosis | Reduced number of atypical melanocytes |
Diffuse irreversible hyperpigmentation (bronzing) (see eFig. 109-2.3) | Increased number of DOPA-positive melanocytes and increased melanin content per unit area and increased number of dermal melanophages |
Wrinkling | |
Fine surface lines | None detected |
Deep furrows (see Figs. 109-2 and 109-3) | Contraction of septae in the subcutaneous fat |
Stellate pseudoscars (see eFig. 109-2.4) | Absence of epidermal pigmentation, altered fragmented dermal collagen |
Elastosis (fine nodularity and/or coarseness) (see Fig. 109-3) | Nodular aggregations of fibrous to amorphous material in the papillary dermis |
Inelasticity | Elastotic dermis |
Telangiectasia | Ectatic vessels often with atrophic walls |
Venous lakes | Ectatic vessels often with atrophic walls |
Purpura (easy bruising) | Extravasated erythrocytes and increased perivascular inflammation |
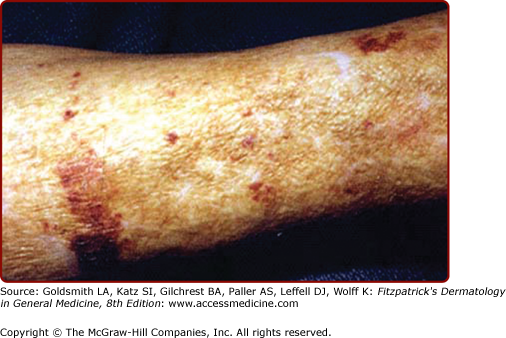
Comedones (maladie de Favre et Racouchot) (see eFig. 109-2.5) | Ectasia of the pilosebaceous follicular orifice |
Sebaceous hyperplasia | Concentric hyperplasia of sebaceous glands |
Figure 109-2
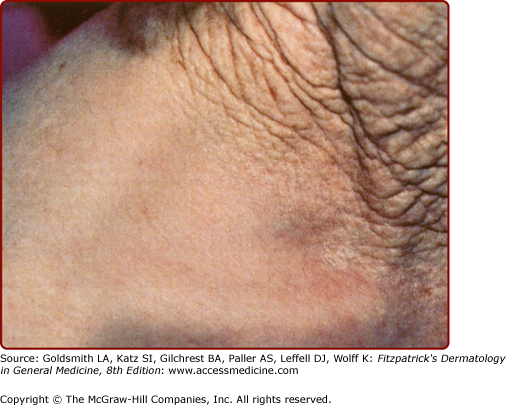
Photoaged versus intrinsically aged skin of an elderly man. Habitually sun-exposed skin above the collar line is prominently wrinkled and lax, in contrast with the equally chronologically aged but sun-protected skin of the lower neck and shoulder. Despite the striking difference in appearance, both areas manifest age-associated functional decrements.
eFigure 109-2.1
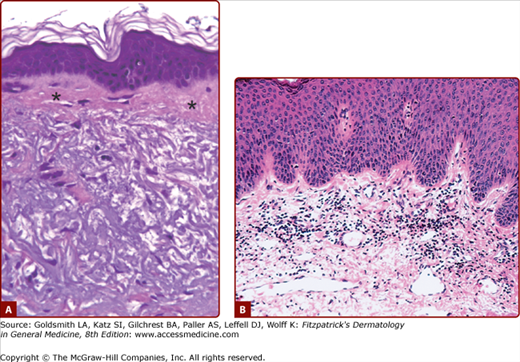
Photodamaged facial skin. A. Large masses of deranged elastic fibers characterize solar elastosis. A thin subepidermal grenz zone (asterisks) is present, and the epidermis is acanthotic. (Used with permission from Jag Bhawan.) B. Marked dermal inflammatory infiltrate associated with the heliodermatitis (dermatoheliosis) that is characteristic of ongoing and chronic actinic exposure. (Used with permission from Lorraine H. Kligman.)
eFigure 109-2.3
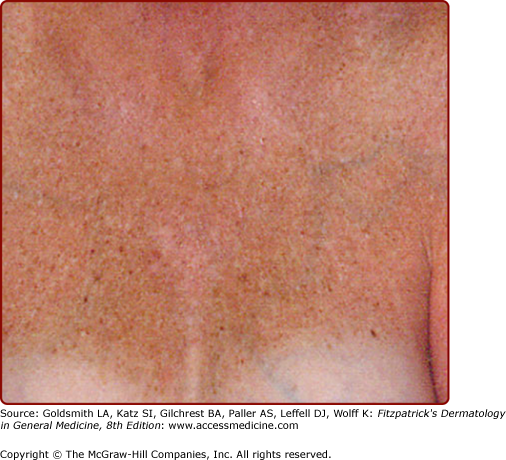
Photoaged skin may be permanently hyperpigmented or “bronzed” as displayed in this 54-year-old woman who remained darkly tanned throughout the year in areas exposed by her sunbathing attire during the summer. (From Yaar M, Gilchrest BA: Ageing and photoageing of keratinocytes and melanocytes. Clin Exp Dermatol 26:583, 2001, with permission.)
eFigure 109-2.5