Fig. 28.1
Transmission electron microscopy of a desmosome and schematic representation of the main molecular components
Genetic disorders affecting the desmosomal assembly, structure or integrity disrupt not only the intercellular adhesion but also the functions in cell communication and regulation, leading to diverse pathologies as cardiomyopathy, epidermal and mucosal blistering, palmoplantar keratoderma and woolly hair [13]. The essential functions of desmosomal proteins in epidermal homeostasis are also reflected by their pathogenic relevance in human infectious and autoimmune disorders and by mouse models [14].
28.2.2 Mouse Models
Mouse models provide invaluable tools to explore the role of proteins during development and adult life, in vivo. However, in the case of desmoplakin and plakoglobin, the human and mice phenotypes do not completely overlap but emphasise the differences between the two species.
The desmoplakin gene was ablated in mice to explore its role in tissue integrity. Homozygous −/− mutant embryos proceeded through implantation, but did not survive beyond embryonic day 6.5. Analysis of these embryos revealed a critical role of desmoplakin, not only in anchoring of intermediate filaments to desmosomes but also in the assembly and stabilisation of desmosomes [15]. While a blastocoel cavity formed and epithelial cell polarity was at least partially established in the desmoplakin (−/−) embryos, the paucity of desmosomal cell-cell junctions severely affected the modelling of tissue architecture and shaping of the early embryo [15].
To analyse the role of plakoglobin during mouse development, the plakoglobin gene was inactivated. Plakoglobin null-mutant embryos died from embryonic day 10.5 onward, due to severe heart defects. Some mutant embryos developed further and died around birth, presumably due to cardiac dysfunction. They demonstrated skin blistering with subcorneal acantholysis. Ultrastructural analysis revealed that desmosomes were greatly reduced in number and structurally altered [16]. Genetic ablation of the plakoglobin gene (jup) in cardiomyocytes in mice led to arrhythmogenic cardiomyopathy similar to Naxos disease in humans [17]. Jup mutant mice generated by inactivating jup in keratinocytes largely recapitulated the clinical features of human palmoplantar keratoderma. They also suffered from skin ulceration and inflammation. Ultrastructural analyses revealed the disruption of the assembly of desmosomes and adherens junctions in jup mutant epidermis [18].
28.3 Acantholytic EB: Clinical and Molecular Features
28.3.1 Clinical Features of Acantholytic EB Associated with Desmoplakin Mutations
All four patients with desmoplakin mutations showed widespread skin erosions already at birth. The denuded skin areas healed without scarring but with residual erythema, and blistering recurred rapidly (Fig. 28.2a). Hair was completely absent, and the nail beds were present but without nails. One patient had mild syndactyly of the fingers and clinodactyly of the fifth finger [3]. The mucosae were severely affected with erosions of the oral cavity, larynx and conjunctivae. Malformation of the ears was also reported with mild unravelling of the superior helices and retroversion [3]. The infants suffered from extensive fluid loss and intubation was necessary in all cases. Cardiac echocardiograms and chest X-rays showed no evidence for heart affliction in three out of four cases, whereas in one case, foetal ultrasound revealed a ventricular hypertrophy with reduced contractility [3]. Ultrasound of the abdomen did not reveal any abnormalities. Nevertheless, all patients died within the first days of life, probably due to multiorgan failure with secondary heart failure precipitated by immense fluid supplementation [2–4]. Postmortem in one case revealed heart dilatation possibly because of overfilling; on microscopy there were no signs of cardiomyopathy [2].
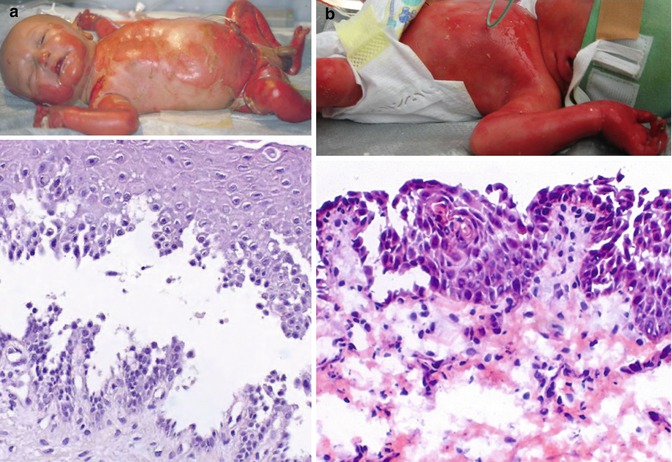
Fig. 28.2
Acantholytic EB: clinical and morphological features of the skin. (a) The upper panel shows extensive erosions in a patient with DSP mutations. In the lower panel, note low suprabasal cleavage and acantholysis. (b) The patient homozygous for the JUP nonsense mutation is shown in the upper panel; the lower panel shows loss of the upper epidermal layers and loss of intercellular adhesion
28.3.2 Clinical Features of Acantholytic EB Associated with Plakoglobin Mutations
Until now, only one patient with acantholytic EB and plakoglobin mutations has been reported in the literature [5]. The disease was coined lethal congenital EB to emphasise the dramatic outcome. The infant presented already at birth with extensive superficial erosions over the entire integument, however without mucosal involvement (Fig. 28.2b). Some areas of the skin seemed to reepithelialise; however, the upper epidermal layers detached extremely easily again. The scalp hair was absent and onycholysis was present. The patient necessitated massive fluid substitution to maintain fluid balance and cardiac function. Electrocardiograms and ultrasound disclosed no abnormalities of the heart or other internal organs. The infant developed sepsis subsequent to Aspergillus fumigatus infection of the skin erosions and died at postnatal day 12 due to respiratory failure.
28.3.2.1 Skin Morphological Features of Acantholytic Epidermolysis Bullosa
In acantholytic EB with DSP mutations, histopathology demonstrates suprabasal skin cleavage and acantholysis of the spinous layer, with intact basal layer (Fig. 28.2a). Transmission electron microscopy may reveal normal or reduced number and size of desmosomes, absent inner dense plaques and disconnection of intermediate filament insertion depending on the remaining length of the truncated molecule as a consequence of the disease-causing mutations. As a result of protein truncation, immunofluorescence staining with domain-specific antibodies to desmoplakin may demonstrate positive or strongly reduced staining with antibodies to the N-terminus and negative staining with antibodies to the C-terminus [3].
In the single patient with a homozygous nonsense plakoglobin mutation leading to loss of protein expression, cell-cell junctions were disrupted throughout all epidermal layers, and the upper layers were lost on long stretches (Fig. 28.2b). Transmission electron microscopy demonstrated loss of desmosomes, with only few desmosomal remnants present. Immunostaining for plakoglobin, desmoplakin and desmoglein 3 was negative, whereas plakophilin 1 was diffusely distributed within the cytoplasm of the keratinocytes [5].
28.3.2.2 Spectrum of Desmoplakin and Plakoglobin Mutations
In the case of desmoplakin and plakoglobin, the consequences of mutations are complex, as are genotype-phenotype correlations. Various mutations lead to different phenotypes, with acantholytic EB being by far the most severe disorder associated with loss-of-function mutations.
Monoallelic dominant DSP mutations cause striate palmoplantar keratoderma II (MIM#612908), whereas recessive mutations were reported in patients with skin fragility-woolly hair syndrome (MIM#607655), arrhythmogenic right ventricular dysplasia 8 (MIM#607450) and dilated cardiomyopathy with woolly hair and keratoderma (Carvajal syndrome, MIM#605676) [13]. The latter was due to a homozygous deletion which produces a premature stop codon leading to a truncated desmoplakin protein missing the C domain of the tail region. A dominant in-frame insertion in the N-terminus of plakoglobin was associated with arrhythmogenic right ventricular dysplasia 12 (MIM#611528) [19], and homozygosity for a two-base pair deletion causes palmoplantar keratoderma, woolly hair and arrhythmogenic right ventricular dysplasia/cardiomyopathy (Naxos disease, MIM#601214). Mild cutaneous disease was reported in patients with biallelic truncating mutations in the N-terminus of plakoglobin [20].
Related posts:
 Kindlin-1 and Its Role in Kindler Syndrome
Kindlin-1 and Its Role in Kindler Syndrome
 Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
Cyclophosphamide in Autoimmune Blistering Diseases: Safety, Efficacy and Evidence Base
Management of Bullous Systemic Lupus Erythematosus
 Azathioprine
Azathioprine
 COL7A1 and Its Role in Dystrophic Epidermolysis Bullosa
COL7A1 and Its Role in Dystrophic Epidermolysis Bullosa
 How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
How to Take a Skin Biopsy Correctly to Diagnose Epidermolysis Bullosa and Autoimmune Bullous Diseases
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


