Differential Diagnosis.
Subepidermal calcified nodules may be confused with verruca vulgaris and pilomatrixomas. On the eyelids they may be confused, in older patients, with xanthelasma.
Treatment.
Surgical excision is the treatment of choice.
Idiopathic Calcinosis of the Scrotum/vulva
Calcinosis of the scrotum is a rare disease characterized by calcified nodules of the scrotal wall. Most patients present between 20 and 40 years of age, but patients as young as 5 years old have been reported [3].
Pathogenesis.
The pathogenesis remains controversial. Many believe that the condition is truly idiopathic. Others maintain that it represents dystrophic calcification of epidermal cysts, foreign bodies, dartoic muscle or eccrine ducts or is secondary to trauma [4]. Idiopathic calcinosis of the vulva may be the female counterpart [5].
Histopathology.
Deposits of calcium are seen either as solitary large nodular masses or multiple small deposits scattered throughout the dermis. There is often a band of compressed collagen and fibrous tissue surrounding the deposits. A true epithelial cyst wall is extremely unusual. There is a variable degree of inflammatory infiltration and foreign body reaction.
Clinical Features.
Firm painless yellowish nodules on the scrotal wall ranging from pinhead to walnut size are seen, numbering between 1 and over 100. Associated symptoms include pruritus and a sensation of scrotal heaviness. The nodules may ulcerate and discharge a cheese-like material.
Differential Diagnosis.
Idiopathic calcinosis of the scrotum is usually misdiagnosed clinically as epidermal cysts.
Treatment.
Full excision or ‘pinch–punch’ excision , mainly for cosmetic reasons, is the treatment of choice [6]. Recurrences are rare.
Milia-Like Idiopathic Calcinosis Cutis
In milia-like idiopathic calcinosis cutis, multiple small 1–2 mm lesions clinically resembling milia develop during childhood. They occur most often on the dorsum of the hands, although they may be seen on face, knees, elbows and soles. An association with Down syndrome has been reported in two-thirds of patients [7,8]. In a few cases they may represent calcification of syringomas. A perforating variety has been described occurring on the pubic and groin area of healthy children [9]. In rare instances, patients with pilomatricomas present with a clinical picture of milia-like calcinosis cutis. Pilomatricomas may heal spontaneously before adulthood, with or without scarring.
Tumoral Calcinosis
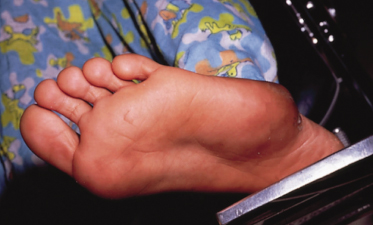
Tumoral calcinosis is a rare disease characterized by the development of large multiple calcified periarticular nodules [10,11]. Tumoral calcinosis may be primary, not associated with any systemic disease (Fig. 95.2), or secondary, associated with some underlying condition such as chronic renal failure, hyperparathyroidism, hypervitaminosis D, sarcoidosis and milk–alkali syndrome and therefore represent a form of metastatic calcification. Primary tumoral calcinosis can be sporadic or familial with an autosomal recessive inheritance. Familial tumoral calcinosis (FTC) is further subdivided into normophosphataemic (OMIM610456) and hyperphosphataemic (OMIM211900) types [12]. Although these two disorders were initially considered part of a clinical continuum they represent distinct entities. Hyperphosphataemic FTC has been shown to result from mutations in three genes: fibroblast growth factor-23 (FGF23), coding for a potent phosphaturic protein; KL encoding Klotho, which serves as a co-receptor for FGF23; and GALNT3, which encodes a glycosyltransferase responsible for FGF23 O-glycosylation [10,11,13–18]. Defective function of any one of these three proteins results in hyperphosphataemia and ectopic calcification. Normophosphataemic FTC is has been linked to mutations in the SAMD9 gene which encodes a putative tumour suppressor and anti-inflammatory protein, and is upregulated by tumour necrosis factor α (TNF-α) [19].
Fig. 95.2 Primary tumoral calcinosis without an identifiable defect in calcium or phosphorus metabolism.
Pathogenesis.
In tumoral calcinosis the calcium is deposited as calcium pyrophosphate crystals. In the hyperphosphataemic variant, increased renal reabsorption of phosphate independent of PTH leads to hyperphosphataemia and therefore tumoral calcinosis may represent a form of metastatic calcification with trauma as a precipitating factor. However, unlike other disorders of metastatic calcification, other organs are not involved. The pathogenesis of the normophosphataemic variant is more obscure.
Histopathology.
Histopathologically, there is a multilocular structure containing solid calcified material and chalky pasty fluid surrounded by a fibrous wall and a foreign body granuloma reaction. The calcified masses are mainly composed of calcium hydroxyapatite with amorphous calcium carbonate and calcium phosphate.
Clinical Features.
Hyperphosphataemic FTC has been mainly reported in Africa and the Middle East [11,20]. Calcified nodules usually develop during the first or second decade of life [21] although they may develop in infancy [22]. Slowly growing calcified masses develop mainly at periarticular locations, with a predilection for skin areas overlying large joints. The hips are most often involved. The lesions may be solitary but are usually multiple. These calcified masses are initially asymptomatic and are often incidentally diagnosed in patients undergoing radiographical investigation for unrelated reasons. The nodules progressively increase in size to reach several centimetres and interfere with movements or cause compression neuropathy. The overlying skin is usually normal, but ulceration, draining sinuses and dermal calcinosis cutis may occur. In some affected individuals, extracutaneous signs may predominate. Dental abnormalities,including hypoplasia and pulp calcifications, may be a prominent feature in some families [18,23]. Angioid streaks and corneal calcifications, as well as testicular microlithiasis, have been described [24]. Hyperphosphataemic FTC has been reported in assocaition with pseudo-xanthoma elasticum (PXE) [25]. The prognosis of the disease is good although patients often undergo many surgical procedures to remove the calcified tumours [20].
Normophosphataemic FTC is less prevalent than the hyperphosphataemic variant [19,26]. Calcified tumour formation is generally preceded by a vasculitis-like rash during the first year of life and is associated with inflammatory manifestations mostly evident in mucosal tissues [21]. This eruption heralds the progressive, but often rapid, development of small, acrally located calcified nodules, which very often ulcerate.
Tumoral calcinosis appears radiologically as dense multilocular calcific masses without bony or joint abnormalities.
Laboratory Studies.
In the normophosphataemic type, serum concentrations of calcium and phosphate are normal whereas in the hyperphosphataemic type, the serum calcium concentration is normal but the phosphate concentration is slightly high. Serum 1,25-dihydroxyvitamin D may be elevated or inappropriately normal, PTH levels are low to low–normal and the circulating FGF23 level are increased. Subtle biochemical abnormalities may be seen in heterozygous carriers of hyperphosphataemic FTC [27].
Treatment.
Surgical excision is the primary form of treatment although recurrences are frequent. Phosphate deprivation and oral aluminium hydroxide administration have been met with some success. Acetazolamide and sevelamer hydrochloride, a non-calcium phosphate binder, have been used in a few patients [17].
References
1 Evans MJ, Blessing K, Gray ES. Subepidermal calcified nodule in children: a clinicopathologic study of 21 cases. Pediatr Dermatol 1995;12:307–10.
2 Afzal MN, Dancea S, de Nanassy J. Mucosal calcified nodule of the hard palate in an infant: case report and review of the literature. Pediatr Pathol Lab Med 1997;17:611–15.
3 Shapiro L, Platt N, Torres-Rodriguez VM. Idiopathic calcinosis of the scrotum. Arch Dermatol 1970;102:199–204.
4 Shah V, Shet T. Scrotal calcinosis results from calcification of cysts derived from hair follicles: a series of 20 cases evaluating the spectrum of changes resulting in scrotal calcinosis. Am J Dermatopathol 2007;29:172–5.
5 Bernardo BD, Huettner PC, Merritt DF, Ratts VS. Idiopathic calcinosis cutis presenting as labial lesions in children: report of two cases with literature review. J Pediatr Adolesc Gynecol 1999;12:157–60.
6 Chang CH, Yang CH, Hong HS. Surgical pearl: pinch–punch excisions for scrotal calcinosis. J Am Acad Dermatol 2004;50:780–1.
7 Becuwe C, Roth B, Villedieu MH et al. Milia-like idiopathic calcinosis cutis. Pediatr Dermatol 2004;21:483–5.
8 Schepis C, Siragusa M, Palazzo R, Batolo D, Romano C. Perforating milia-like idiopathic calcinosis cutis and periorbital syringomas in a girl with Down syndrome. Pediatr Dermatol 1994;11:258–60.
9 Eng AM, Mandrea E. Peforating calcinosis cutis presenting as milia. J Cutan Pathol 1981;8:247–50.
10 Joseph L, Hing SN, Presneau N et al. Familial tumoral calcinosis and hyperostosis-hyperphosphataemia syndrome are different manifestations of the same disease: novel missense mutations in GALNT3. Skeletal Radiol 2010;39:63–8.
11 Sprecher E. Familial tumoral calcinosis: from characterization of a rare phenotype to the pathogenesis of ectopic calcification. J Invest Dermatol 2010;130:652–60.
12 Smack D, Norton SA, Fitzpatrick JE. Proposal for a pathogenesis-based classification of tumoral calcinosis. Int J Dermatol 1996;35:265–71.
13 Araya K, Fukumoto S, Backenroth R et al. A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J Clin Endocrinol Metab 2005;90:5523–7.
14 Barbieri AM, Filopanti M, Bua G, Beck-Peccoz P. Two novel nonsense mutations in GALNT3 gene are responsible for familial tumoral calcinosis. J Hum Genet 2007;52:464–8.
15 Chefetz I, Heller R, Galli-Tsinopoulou A et al. A novel homozygous missense mutation in FGF23 causes familial tumoral calcinosis associated with disseminated visceral calcification. Hum Genet 2005;118:261–6.
16 Garringer HJ, Mortazavi SM, Esteghamat F et al. Two novel GALNT3 mutations in familial tumoral calcinosis. Am J Med Genet A 2007;143A:2390–6.
17 Lammoglia JJ, Mericq V. Familial tumoral calcinosis caused by a novel FGF23 mutation: response to induction of tubular renal acidosis with acetazolamide and the non-calcium phosphate binder sevelamer. Horm Res 2009;71:178–84.
18 Specktor P, Cooper JG, Indelman M, Sprecher E. Hyperphosphatemic familial tumoral calcinosis caused by a mutation in GALNT3 in a European kindred. J Hum Genet 2006;51:487–90.
19 Chefetz I, Ben AD, Browning S et al. Normophosphatemic familial tumoral calcinosis is caused by deleterious mutations in SAMD9, encoding a TNF-alpha responsive protein. J Invest Dermatol 2008;128:1423–9.
20 Carmichael KD, Bynum JA, Evans EB. Familial tumoral calcinosis: a forty-year follow-up on one family. J Bone Joint Surg Am 2009;91:664–71.
21 Metzker A, Eisenstein B, Oren J, Samuel R. Tumoral calcinosis revisited: common and uncommon features – report of ten cases and review. Eur J Pediatr 1988;147:128–32.
22 Polykandriotis EP, Beutel FK, Horch RE, Grünert J. A case of familial tumoral calcinosis in a neonate and review of the literature. Arch Orthop Trauma Surg 2004;124:563–7.
23 Frishberg Y, Topaz O, Bergman R et al. Identification of a recurrent mutation in GALNT3 demonstrates that hyperostosis-hyperphosphatemia syndrome and familial tumoral calcinosis are allelic disorders. J Mol Med 2005;83:33–8.
24 Campagnoli MF, Pucci A, Garelli E et al. Familial tumoral calcinosis and testicular microlithiasis associated with a new mutation of GALNT3 in a white family. J Clin Pathol 2006;59:440–2.
25 Mallette LE, Mechanick JI. Heritable syndrome of pseudoxanthoma elasticum with abnormal phosphorus and vitamin D metabolism. Am J Med 1987;83:1157–62.
26 Topaz O, Indelman M, Chefetz I et al. A deleterious mutation in SAMD9 causes normophosphatemic familial tumoral calcinosis. Am J Hum Genet 2006;79:759–64.
27 Ichikawa S, Lyles KW, Econs MJ. A novel GALNT3 mutation in a pseudoautosomal dominant form of tumoral calcinosis: evidence that the disorder is autosomal recessive. J Clin Endocrinol Metab 2005;90:2420–3.
Dystrophic Calcification
Dystrophic calcification has been reported in a variety of disorders (Table 95.1).
Connective Tissue Diseases
Cutaneous calcification can occur in all connective tissue diseases. The mechanism for dystrophic calcification in connective tissue diseases is unknown [1].
Dermatomyositis is the disorder most frequently associated with calcification in children (Fig. 95.3). Between 50% and 70% of children with dermatomyositis will develop calcinosis compared to 20% of adults [2]. A high incidence of staphylococcal infection in children with dermatomyositis who subsequently develop calcinosis has been noted [3]. Calcinosis cutis tends to develop 2–3 years after the onset of dermatomyositis, and rarely is the presenting sign. Calcifications may range from small asymptomatic skin nodules which rarely interfere with function (calcinosis circumscripta) to large tumoral masses in the muscles or sheet-like deposits in the intermuscular fascial planes (calcinosis universalis). In a few children, often those with erythroderma and diffuse cutaneous vasculitis from the disease onset, the calcification may be severe resembling an ‘exoskeleton’. Nodular calcium deposits are seen primarily in the muscular groups most severely involved (i.e. those of the shoulder and pelvic girdles, followed by the elbows and knees). Episodes of ulceration, calcium extrusion and cellulitis accompanied by severe systemic symptoms are common. After expulsion of calcium, the ulcerations usually heal promptly. Calcium deposits increase over a period of several months, after which they remain stable for a few more months and then improve slowly. The degree of calcinosis seems to correlate with the degree of muscular involvement, a longer time to diagnosis and treatment (and longer disease duration) [2,4]. Generally, calcinosis develops in the most seriously ill patients who survive. For this reason, although it can be severely incapacitating, it is viewed as a good prognostic sign for survival. Recently, children with autoantibodies to a 140-kDa protein have been shown to have a higher incidence of calcinosis [5]. B-cell lymphoma has been described to arise in a foci of calcinosis in a girl with dermatomyositis [6]. Calcinosis is also frequent in scleroderma and CREST syndrome (calcinosis, Raynaud phenomenon, oesophageal involvement, sclerodactyly and telangiectasia), an association that has been recognized as the Thibierge–Weissenbach syndrome. Calcification occurs in 27% of patients with acrosclerosis and develops an average of 10 years after the disease onset, and therefore is not commonly seen during childhood. Calcium deposits are usually more limited than in dermatomyositis, and calcinosis universalis is rare. The sites of predilection are the hands (finger fat pads) and upper extremities, in areas of sclerosis, and periarticular areas but it can be found elsewhere [7]. Paraspinal and intraspinal calcifications may be seen [8]. Cutaneous calcification, although rare, may occur in morphoea and linear scleroderma [9].
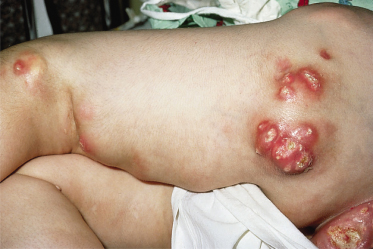
Calcinosis is less common in systemic subacute cutaneous and chronic cutaneous lupus and is extremely rare in children [10]. The lesions may be located underneath the cutaneous lesions of lupus or elsewhere. Calcification usually develops an average of 8 years after the disease onset, although it may also precede other symptoms.
Dystrophic calcinosis in connective tissue disease runs an independent course from that of the underlying disease. However, there is some evidence that aggressive management of dermatomyositis results in decrease incidence of calcinosis [4]. Several forms of treatment have been attempted, including diets high in phosphate and low in calcium, calcium-chelating agents, such as disodium ethylenediamine tetra-acetic acid (EDTA) and diphosphonates and oral aluminium hydroxide, which is a phosphate chelator [1]. These treatments are associated with significant side-effects and in general have yielded unimpressive results. In a few patients, probenecid and colchicine have appeared to be beneficial. Warfarin has been shown to decrease the tissue levels of the calcium-binding amino acid, γ-carboxyglutamic acid, by inhibition of the vitamin K-dependent γ-carboxylase [11]. Low-dose warfarin only improves the findings on bone scans without any clinically significant effect. Diltiazem has been effective in a few patients [12]. The presumed mechanism of action is its inhibitory effect on calcium transport into the cells. Treatment of the underlying disease with intraveous immunoglobulin has led to conflicting results [13,14]. Intralesional administration of steroids may be beneficial in localized deposits. Surgical removal of the calcium deposits is indicated when deposits are large, painful or subjected to frequent ulceration. Postoperative recurrences are possible. Spontaneous regression of calcinosis has been described.
Panniculitis
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


