Fig. 50.2 Discrete, lentil-sized, flat papules are visible on the buttocks and limbs of this adolescent, while the trunk is spared.
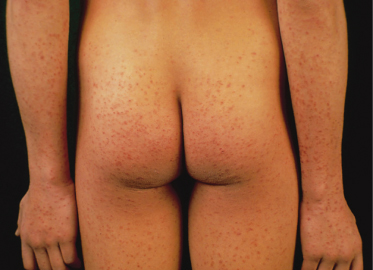
Fig. 50.3 This infant presents papular and purpuric lesions on the cheeks, together with similar lesions on the buttocks and limbs.
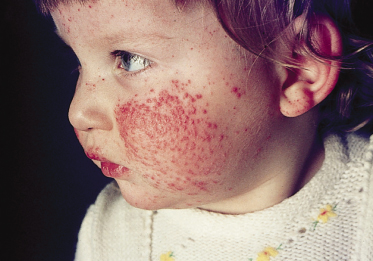
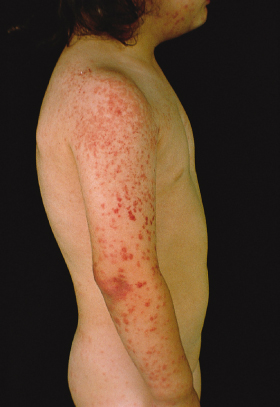
Fig. 50.4 Purpuric lesions as a result of capillary fragility can be provoked when a touniquet is applied for venepuncture as in this young patient.
The Gianotti–Crosti syndrome usually begins on the thighs and buttocks, then spreads to the extensor aspects of the arms and finally involves the face. The simultaneous appearance of lesions in all of these anatomical areas can also be observed. The rash develops within a week. The individual lesions are hemispherical, a few millimetres in diameter (lesions of 8–10 mm in diameter are uncommon) and their colour varies from rose to red–brown. The hue may be modified by purpuric staining, especially on the legs, in areas of trauma and on the face after prolonged crying. The eruption fades with mild desquamation in 3–4 weeks. A longer disease course, of up to 6–8 weeks, is uncommon [4,53,54].
Recurrences are exceptional [32]. Constitutional symptoms are usually mild; low-grade fever and lassitude sometimes occur. Pruritus is not a prominent feature, and excoriations are never seen. Lymphadenopathy is common but does not occur in every case. Enlarged nodes are most often located in the axilla and inguinal region. They are soft and freely movable. Lymphadenopathy can persist for weeks after the rash.
In hepatitis B forms, the liver is usually enlarged but not tender; jaundice is exceptional. Serious complications are very rare, but some patients affected by the hepatitis B form have shown histological evidence of chronic periportal hepatitis [13]. Chronic active hepatitis is extremely uncommon [10]. The syndrome, like most other viral exanthems, mainly affects children of preschool age. Occasional adult cases have occurred [55]. In the author’s series [24] of more than 300 patients, the age of onset was between 6 months and 14 years (mean 2 years) and the syndrome was only slightly more frequent in boys. The eruption is more common in spring and early summer [24,53,56]. An association with atopy [57] and hyperimmunoglobulin E syndrome [58] have been also described.
Laboratory findings are inconstant. There may be a leucopenia or a slight leucocytosis with increased monocytes; the erythrocyte sedimentation rate is not raised. In hepatitis B-positive cases, there are high serum levels of hepatic enzymes (e.g. glutamicoxaloacetic transaminase, glutamic–pyruvic transaminase, aldolase, lactic dehydrogenase, alkaline phosphatase). Glutamicoxaloacetic transaminase and glutamic–pyruvic transaminase titres can be higher than 1000 or 2000 U/mL. The hepatitis begins at the same time as, or 1–2 weeks after, the onset of the dermatitis. The change in serum enzymes usually persists for 3–6 months. In hepatitis B-associated cases, viral markers are detectable in the serum at the time as the syndrome is noted. Subtyping of hepatitis B surface antigen has consistently revealed the ayw [15] or adr [16] determinants. Some of the other viruses that cause Gianotti–Crosti syndrome (e.g. Epstein–Barr) may also cause mild hepatitis. In hepatitis B-negative cases, hepatomegaly and liver function abnormalities, if present, are slight. Serum transaminases rarely exceed 100 U/mL.
Prognosis.
Prognosis is good. Severe active liver disease in patients with hepatitis B may regress without treatment [7,15]. One case with Gianotti–Crosti syndrome secondary to EBV was associated with Bell’s palsy [59]. Fatal outcome has been reported only once [60].
Differential Diagnosis.
In most cases, the morphology and distribution of skin lesions are distinctive. Conditions to be differentiated from Gianotti–Crosti syndrome are Henoch–Schönlein purpura, erythema multiforme, hand–foot–mouth disease, the acrolocated form of pityriasis lichenoides [4,29,49] and, to a lesser extent, the recently described asymmetrical periflexural exanthem of childhood [61]. In Henoch–Schönlein purpura, the lesions are larger and are rarely papular; they tend to occur in successive crops. The patients are generally older, and may develop joint tenderness and visceral symptoms. In erythema multiforme, which is also rare in young children, the lesions are larger and more oedematous, and are often dusky and targetoid. Mucous membranes may be involved. Hand–foot–mouth disease may occur on the knees, elbows and buttocks, and mimic Gianotti–Crosti syndrome. However, mucosal lesions are usually observed in the first stage of the eruption. Palmar–plantar vesicular lesions of hand–foot–mouth disease are characteristic and persistent. In the acrolocated form of pityriasis lichenoides, the lesions appear in successive crops, and have a darker, brownish colour and central scale. This disease runs a much longer course. In rare cases, the lesions of Gianotti–Crosti syndrome are not manifestly symmetrical, and must be differentiated from those of the asymmetrical periflexural exanthem of childhood. In this disease, however, lesions appear first on one side of the body and spread in a minor proportion of cases to the other side; the face is always spared. Interestingly, this differential diagnosis was proposed over 30 years ago by Gianotti and Crosti [4], only 1 year after the first description of the periflexural exanthem by Brunner et al. [62]. In rare cases, Gianotti–Crosti syndrome must be differentiated from papular urticaria and frictional lichenoid dermatitis; in adults, lichen planus and lichenoid drug eruptions should be considered. A complete list of differential diagnoses is illustrated in Table 50.3.
Table 50.3 Gianotti–Crosti syndrome: differential diagnosis
| Common |
| Henoch–Schoenlein purpura |
| Erythema multiforme (early) |
| Hand–foot–mouth disease |
| Asymmetrical periflexural exanthem of childhood |
| Lichen ruber planus (especially adults) |