In the stratum granulosum (SG), the large stores of proteins and lipids produced during differentiation are visible and form the keratohyalin granules and lamellar bodies (LB). Upon transition from the SG to the SC, the contents of these granules are released, resulting in the remodelling of the keratinocytes into corneocytes, a process termed cornification. This transition is accompanied by the loss of their intracellular organelles and nuclei. The proteins derived from the keratohyalin granules reinforce the plasma membrane, forming an insoluble layer referred to as the cornified envelope (CE) [reviewed by [1]. The CE is mainly composed of loricrin, involucrin, filaggrin and small proline rich proteins (SPRs), which are cross-linked together by the action of transglutaminases. Filaggrin also aggregates the keratin fibres of the cellular cytoskeleton into bundles, collapsing the corneocytes into flattened discs with a large surface area (approximately 1000 µm2 for mature surface corneocytes). The resulting corneocytes have a geometrical morphology and interlock together in layers of approximately 20 corneocytes deep, spanning between 10 and 20 µm, depending on the body site [3]
The majority of filaggrin protein does not persist beyond the deepest two layers of the SC [4]. As the corneocytes mature, filaggrin is extensively deiminated through the actions of the enzyme peptidyl-deiminase to reduce keratin-binding affinity, and subsequently degraded into small peptides and then free amino acids. The free amino acids are then catabolized into the constituents of natural moisturizing factor (NMF), including sodium pyrrolidone carboxylic acid (NaPCA) and trans-urocanic acid (tUCA); NMF also contains lactic acid and urea from other prescursors [4]. NMF is essential for the retention of water within corneocytes and results in their optimal hydration and tumidity. NaPCA and lactic acid, in particular, are intensely hydroscopic; they both absorb water and dissolve in their own water, acting as very efficient humectants, which leads to a swelling of corneocytes, preventing the development of gaps between them.
To help prevent the loss of water from the swollen corneocytes, they are surrounded by lipid lamellae, crystalline substances composed of ceramides, cholesterol, fatty acids and cholesterol esters [5]. The lipid lamellae are extruded into the extracellular space between the corneocytes from the secretory LB, which are thought to be branched tubular structures extending from the granular keratinocyte trans-Golgi network [6]. In addition to preventing water loss, the lipid lamellae restrict the penetration of water-soluble materials. The CE acts as a scaffold for the attachment of lipids, including ceramides, from the lamellar matrix, which aggregate to form the lipid envelope [1]. The corneocytes are therefore enmeshed in what is believed to be a single and coherent lamellar gel that confers flexibility to the barrier.
The intercellular desmosomal junctions connecting the corneocytes together are also reinforced following the association with corneodesmosin (CDSN). The incorporation of corndeodesmosin marks the transition from desmosome to corneodesmosome [7]. This locks the corneocytes together, providing tensile strength for the SC to resist shearing forces. Corneodesmosin is a 52 kDa protein, packaged in LB and secreted into the extracellular space at the transition between the SG and SC [8]. LB also deliver a cocktail of proteases which progressively break down the corneodesmosomal junctions as the corneocytes mature. This results in the disassociation of mature corneocytes (or squames) in the uppermost layers of the SC. This process of corneocyte shedding, known as desquamation, counterbalances the generation of new keratinocytes in the basal layer so that the whole epidermis is continually renewed [9].
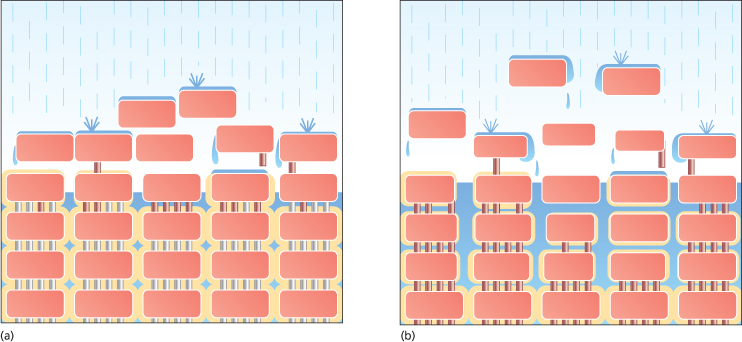
The primary barrier to the penetration of irritants and allergens through the skin, the skin barrier, is located in the lower part of the SC. This is the point at which the integrity of the barrier is greatest, where cornification culminates prior to its ensuing breakdown. Elias described this barrier as being like a brick wall, where the corneocytes are analogous to bricks and the lipid lamellae to cement sealing the bricks together [10]. Extending this model, Cork et al. likened the corneodesmosomes to iron rods that pass down through the bricks to add strength to the wall (Fig. 27.2) [11]. Desquamation can be envisaged as rusting of these iron rods in the upper layers of the brick wall.
Fig. 27.2 The ‘brick wall’ analogy of the skin barrier [14,15]. The wall represents the barrier to the loss of water and to the penetration of irritants/allergens. It is made up of bricks, representing the corneocytes, held together by iron rods, representing the corneodesmosomal junctions, and surrounded by mortar, representing the lipid lamellae. (a) The intact ‘healthy’ skin barrier. The rusting of the iron rods in the uppermost layers represents desquamation. (b) The defective skin barrier present in AD. The broad skin barrier defect described in the text can be visualized as a broken wall built with an insufficient amount of poor-quality mortar. There is an increased rate of desquamation represented by the increased rusting of the iron rods. The poor construction leads to the development of cracks that permit the entry of irritants and allergens.
Desquamation
Desquamation involves a network of degradatory proteases, regulated by protease inhibitors, which break down the extracellular corneodesmosome adhesion proteins securing the corneocytes together [9]. This process is largely attributed to the kallikrein-related peptidases (KLK), a family of serine proteases possessing either trypsin-like or chymotrypsin-like activity. To date, seven trypsin-like KLKs, KLK5 (previously SCTE), KLK6, KLK8, KLK10, KLK11, KLK13 and KLK14, and one chymotrypsin-like KLK, KLK7 (previously stratum corneum chymotryptic enzyme, SCCE), have been specifically identified in the SC [12]. At least a further three, including KLK1, KLK3 and KLK9, have been found in total skin extracts. Of these, KLK5 has been shown to hydrolyse DSG, DSC and CDSN, whereas KLK7 cleaves DSC and CDSN only. KLK6 and KLK14, exhibiting the highest level of activity, are also capable of cleaving DSG.
All of the KLKs are expressed as inactive precursors, whereby removal of pro-peptides by trypsin digestion is required for the formation of proteolytically active enzymes [12]. The ability of the KLK with trypsin-like activity to activate other family members, coupled with their different levels of expression and activity within the SC, creates a complex system of hierarchical activation. KLK5, which is capable of self-activation, is thought to trigger this proteolytic cascade, ultimately leading to the breakdown of corneodesmosomes. Other enzymes found in the SC and capable of degrading corneodesmosomal adhesion proteins include the cysteine proteases cathepsin L2 (stratum corneum thiol protease) and stratum corneum L-like enzyme [13], and the aspartate protease cathepsin D [14].
The activity of this proteolytic network is strictly regulated by a number of protease inhibitors, including the pH-dependent lymphoepithelial kazal-type 5 serine protease inhibitor (LEKTI) [15]. LEKTI is composed of 15 potential serine proteinase inhibitory domains, at least four of which have confirmed activity against members of the kallikrein family, including KLK5, KLK6, KLK7 and KLK14. It is expressed by granular cells and delivered by LB vesicles to the superficial SG layers, ahead of its target proteases, prohibiting undesired proteolytic activity in the lower layers of the SC [8]. This is achieved owing to the highly organized and compartmentalized nature of LB trafficking. At the SG–SC interface. LEKTI is co-localized with KLKs in the extracellular space where the pH is near neutral. Under these conditions, LEKTI was found to be a potent inhibitor of both KLK5 and KLK7 [15]. As the pH becomes more acidic, the inhibitory potential of LEKTI is reduced. In the superficial layers of the SC, inhibition by LEKTI is sufficiently reduced to support localized desquamation.
LEKTI, encoded by the SPINK5 gene, is a member of a group of serine protease inhibitors of the Kazal type (SPINK), of which a second member has recently been identified in human skin [14]. Encoded by the SPINK9 gene, LEKTI-2 was found to inhibit KLK5, but not KLK7 and KLK14, proteolytic activity. Human epidermis also expresses serine leucoprotease inhibitor (SLPI) and elafin, otherwise known as skin-derived antileucoprotease (SKALP), which specifically inhibits KLK7, and the cystatin protease inhibitors A, C and M/E, which are specific for cysteine proteases [14]. Cystatin A is also secreted in sweat and forms a protective layer over the surface of the skin against exogenous cysteine proteases such as those produced by house dust mites and Staphylococcus aureus (Staph. aureus) [16].
Stratum Corneum pH
A pH gradient exists across the SC, starting with a pH close to 7 at the SG–SC interface and changing to an acidic pH, of around 5, in the superficial layers of the SC (see Fig. 27.1c) depending on age, sex and the anatomical site tested [17]. This is a simplified view of the spatial distribution of the pH gradient, which in fact occurs through the progressive accumulation of acidic micro-domains. At least three endogenous pathways contribute to SC acidification (Fig. 27.3): the generation of free fatty acids (FFA) from phospholipids via the action of secretory phospholipase A2 (sPLA2) or from ceramide via the action of epidermal ceramidase; the activity of the non-energy dependent sodium-proton exchanger-1 (NHE1); and the degradation of filaggrin into NMF [17–19]. In the latter pathway, filaggrin degradation yields free amino acids including histidine, which is catabolized into tUCA by histidase. The resulting acidic pH of the skin has a strong antimicrobial effect, decreasing skin colonization by pathogenic bacteria and favouring the adhesion of non-pathogenic bacteria to the SC [17].
Fig. 27.3 Stratum corneum pH is centrally involved in the regulation of key skin barrier functions. Histidase (HDase) catalyses the conversion of histidine into trans-urocanic acid (tUCA), a component of NMF. Essential FFA are generated from phospholipids and ceramide by the actions of secretory phospholipase A2 (PLA2) and ceramidase (CDase) respectively. The lipid biosynthetic enzymes β-glucocerebrosidase (βGCase) and acid sphingomyelinase (SMase) are inhibited at high pH, whereas the degradatory serine proteases, including the kallikreins, are activated.
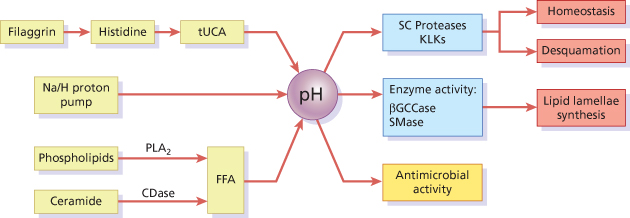
The pH gradient is also vital for maintaining normal barrier function, including the regulation of lipid lamellae biosynthesis and desquamation. The generation of lamellar components relies on the conversion of glycosylceramides and sphingomyelin into ceramides by β-glucocerebrosidase (β-GlucCer’ase) and acid sphingomyelinase, both of which have an acid optimum pH [17]. Furthermore, desquamation is largely dependent on the activity of serine proteases with alkaline pH optima [9], and regulated by the pH-sensitive inhibitor LEKTI [15]. For instance, a change in pH from 5.5 to 7.5 increases KLK7 activity by 50% [9]. When the skin pH is increased by blocking either sPLA2 or NHE1, barrier abnormalities are observed, which can be corrected by co-exposure of inhibitor-treated areas to an acidic buffer [20,21]. Moreover, a delay in skin barrier recovery occurs when the skin is immersed in neutral pH buffers [22]. When hairless mice were treated with ‘superbases’ that neutralize skin surface pH, a rapid activation of serine protease activity was observed, with consequent degradation of corneodesmosomes [23]. This was accompanied by decreased β-GlucCer’ase activity, resulting in incompletely processed lipid lamellae membranes.
References
1 Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol 2005;6(4):328–40.
2 Al-Amoudi A, Frangakis AS. Structural studies on desmosomes. Biochem Soc Trans 2008;36(Pt 2):181–7.
3 Ya-Xian Z, Suetake T, Tagami H. Number of cell layers of the stratum corneum in normal skin – relationship to the anatomical location on the body, age, sex and physical parameters. Arch Dermatol Res 1999;291(10):555–9.
4 Harding CR, Watkinson A, Rawlings AV et al. Dry skin, moisturization and corneodesmolysis. Int J Cosmet Sci 2000;22(1):21–52.
5 Rawlings AV. Trends in stratum corneum research and the management of dry skin conditions. Int J Cosmet Sci 2003;25(1–2):63–95.
6 Norlen L, Al-Amoudi A, Dubochet J. A cryotransmission electron microscopy study of skin barrier formation. J Invest Dermatol 2003;120(4):555–60.
7 Serre G, Mils V, Haftek M et al. Identification of late differentiation antigens of human cornified epithelia, expressed in re-organized desmosomes and bound to cross-linked envelope. J Invest Dermatol 1991;97(6):1061–72.
8 Ishida-Yamamoto A, Simon M, Kishibe M et al. Epidermal lamellar granules transport different cargoes as distinct aggregates. J Invest Dermatol 2004;122(5):1137–44.
9 Caubet C, Jonca N, Brattsand M et al. Degradation of corneodesmosome proteins by two serine proteases of the kallikrein family, SCTE/KLK5/hK5 and SCCE/KLK7/hK7. J Invest Dermatol 2004;122(5):1235–44.
10 Elias PM. Epidermal lipids, barrier function, and desquamation. J Invest Dermatol 1983;80 Suppl:44s–9s.
11 Cork MJ, Robinson DA, Vasilopoulos Y et al. New perspectives on epidermal barrier dysfunction in atopic dermatitis: gene–environment interactions. J Allergy Clin Immunol 2006;118(1):3–21; quiz 2–3.
12 Eissa A, Diamandis EP. Human tissue kallikreins as promiscuous modulators of homeostatic skin barrier functions. Biol Chem 2008;389(6):669–80.
13 Bernard D, Mehul B, Thomas-Collignon A et al. Analysis of proteins with caseinolytic activity in a human stratum corneum extract revealed a yet unidentified cysteine protease and identified the so-called “stratum corneum thiol protease” as cathepsin l2. J Invest Dermatol 2003;120(4):592–600.
14 Meyer-Hoffert U. Reddish, scaly, and itchy: how proteases and their inhibitors contribute to inflammatory skin diseases. Arch Immunol Ther Exp (Warsz) 2009;57(5):345–54.
15 Deraison C, Bonnart C, Lopez F et al. LEKTI fragments specifically inhibit KLK5, KLK7, and KLK14 and control desquamation through a pH-dependent interaction. Mol Biol Cell 2007;18(9):3607–19.
16 Kato T, Takai T, Mitsuishi K et al. Cystatin A inhibits IL-8 production by keratinocytes stimulated with Der p 1 and Der f 1: biochemical skin barrier against mite cysteine proteases. J Allergy Clin Immunol 2005;116(1):169–76.
17 Fluhr J, Bankova LG. Skin surface pH: mechanism, measurement, importance. In: Serup J, Jemec GB, Grove GL (eds) Handbook of Non-Invasive Methods and the Skin. Boca Raton, FL: CRC Press, 2006, pp. 411–20.
18 Nakagawa N, Sakai S, Matsumoto M et al. Relationship between NMF (lactate and potassium) content and the physical properties of the stratum corneum in healthy subjects. J Invest Dermatol 2004;122(3):755–63.
19 Houben E, Hachem JP, de Paepe K et al. Epidermal ceramidase activity regulates epidermal desquamation via stratum corneum acidification. Skin Pharmacol Physiol 2008;21(2):111–18.
20 Behne MJ, Meyer JW, Hanson KM et al. NHE1 regulates the stratum corneum permeability barrier homeostasis. Microenvironment acidification assessed with fluorescence lifetime imaging. J Biol Chem 2002;277(49):47399–406.
21 Fluhr JW, Kao J, Jain M et al. Generation of free fatty acids from phospholipids regulates stratum corneum acidification and integrity. J Invest Dermatol 2001;117(1):44–51.
22 Mauro T, Holleran WM, Grayson S et al. Barrier recovery is impeded at neutral pH, independent of ionic effects: implications for extracellular lipid processing. Arch Dermatol Res 1998;290(4):215–22.
23 Hachem JP, Man MQ, Crumrine D et al. Sustained serine proteases activity by prolonged increase in pH leads to degradation of lipid processing enzymes and profound alterations of barrier function and stratum corneum integrity. J Invest Dermatol 2005;125(3):510–20.
Skin Barrier Homeostasis
Regulation of Skin Barrier Structure and Function
A distinct gradient of calcium ions (Ca2+) exists across the epidermis (see Fig. 27.1b), starting with low levels in the basal layer(s) and progressively increasing towards the upper SG, where it peaks, before sharply declining across the SC [1,2]. Ca2+ regulates the expression of differentiation-dependent genes in keratinocytes, such as those encoding proteins of the CE [3]. As such, the Ca2+ gradient plays a key role in maintaining the different stages of differentiation. Other ions, including potassium, also exhibit specific gradients across the epidermis, which play important roles in regulating barrier homeostasis [4]. The maintenance of these ion gradients across the epidermis is interdependent on the ability of the epidermis, specifically the SC, to act as a permeability barrier [1]. Poor permeability barrier function, defined by a decreased ability to retain water (increased permeability), is accompanied by increased ion flux with consequent perturbation of epidermal ion gradients. Acute barrier damage, by tape-stripping for example, causes rapid ion flux, resulting in the loss of Ca2+ in the SG. This disruption of the Ca2+ gradient triggers rapid secretion of LB contents in the upper SG and restoration of permeability barrier function. The reduced level of Ca2+ in the SG also precludes the expression of differentiation-dependent genes, thereby promoting proliferation [3]. If the permeability barrier is artificially restored using a vapour-permeable membrane barrier, repair does not take place, but the Ca2+ gradient is rapidly restored [1]. Similarly, if exogenous Ca2+ is applied to the skin, the repair process is inhibited [4]. This highlights the role of the Ca2+ gradient in regulating the release of LB contents at the transition between the SG and SC in addition to the barrier repair process following disruption.
Barrier disruption also results in elevation of pH within the uppermost layers of the epidermis (Fig. 27.4), and the subsequent elevation of serine protease activity [5]. Alterations in trypsin-like serine protease activities, including KLK5 and KLK14, play a key role in skin barrier homeostasis through their ability to activate the PAR2 signalling cascade by direct cleavage of PAR2 [5,6]. PAR2 is a member of the protease-activated receptor (PAR) family of G-coupled receptors, involved in innate immune inflammatory responses and pruritus [7]. Its activation, as a result of acute disruption or chronic abrogation of the skin barrier, triggers release of the proinflammatory cytokines interleukin (IL)-8, ICAM-1, TNF-α and thymic stromal lymphopoietin (TSLP) [8]. TSLP, an IL-7-like cytokine, is a mediator of proallergic inflammation [9]. Inducible expression of TSLP in mice results in the development of AD-like lesions [10]. Activation of PAR2 also results in the inhibition of LB secretion and promotion of cornification (terminal differentiation), albeit with a delay of about 30 minutes [11]. This delay permits the secretion of the preformed pool of LB, triggered by extracellular Ca2+ flux. After this initial release of lamellar lipids, PAR2 activation prevents further LB secretion and promotes rapid cornification of the uppermost granular cells. This enhancement of cornification is thought to arise due to the PAR2-triggered increase in intracellular calcium, derived from internal stores, which occurs independently of extracellular Ca2+ levels [12].
Fig. 27.4 The serine protease (SP)-PAR2 pathway (red) and associated mechanisms of regulating barrier homeostasis (blue and green). Environmental factors (left-hand boxes and dashed lines) influence barrier homeostasis, resulting in exacerbated skin barrier breakdown and inhibition of barrier repair.
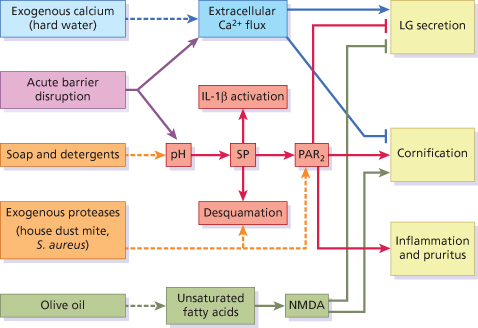
In short, the simultaneous disruption of the Ca2+ gradient and activation of the serine protease-PAR2 pathway following acute barrier disruption results in the fortification of the skin barrier by the co-ordinated and rapid transition of granular cells into corneocytes encased in a preformed lamellar mesh (see Fig. 27.4). Notably, inhibition of the serine protease-PAR2 pathway using serine protease inhibitors was found to facilitate recovery of the permeability barrier following acute disruption [5]. This is in agreement with the opposing effect resulting from activation of this pathway by elevation of SC pH [13]. Activation of the serine protease-PAR2 pathway is therefore detrimental in terms of rapidly repairing permeability barrier function.
Activation of calcium-permeable ionotropic channels expressed by keratinocytes was also found to inhibit barrier recovery [14]. Of these, the NMDA-type glutamate receptor, upon activation by glutamate or the specific agonist NMDA, triggers the influx of calcium into keratinocytes. The topical application of certain unsaturated fatty acids exhibited similar effects, resulting in altered differentiation [14]. This led to the finding that the unsaturated fatty acid oleic acid mediates its effect on the skin barrier through NMDA-type glutamate receptors (see Fig. 27.4). This may be surprising because oleic acid is the main component of olive oil (55–83%), which is widely used topically as a skin protector and softener. However, olive oil, and oleic acid, have been shown to adversely affect the water-retaining properties of the skin, induce scaling and trigger an inflammatory response [14,15]. Not all fatty acids have this effect, and linoleic acid in particular was shown to improve barrier function. This is attributed to its potent activation of perioxisome proliferator-activated receptor α (PPARα) [16]. PPARα is a member of the nuclear hormone family of receptors involved in regulating proliferation, inflammation and barrier homeostasis in response to a range of lipid metabolites.
A greater knowledge of the mechanisms underlying skin barrier homeostasis helps us to understand the effect of the environment on barrier function. For instance, the use of soap and harsh ionic detergents has a profound effect on the surface pH of skin, and thereby significant detrimental effect on skin barrier function (see Fig. 27.4) [17]. Mucke and colleagues reported that washing the skin with soap causes an increase in pH by 3 units on the palms for more than 90 minutes [18]. Increases in skin surface pH, resulting from the use of soap and harsh detergents, cause significant structural damage indicated by a decreased ability of the barrier to retain water [19]. Significant thinning of the SC was also observed following washing with soap, consistent with altered activity of epidermal proteases [20].
Our environment also contains a number of different sources of exogenous proteases with the potential to degrade the skin barrier (facilitate desquamation) and activate PAR2 (see Fig. 27.4). For instance, Staph. aureus releases several serine proteases known as exfoliative toxins, which break down the skin barrier via cleavage of DSG to facilitate colonization [21]. An association with a defective skin barrier is evident from the dramatically elevated colonization of patients suffering from AD with Staph. aureus (more than 90%) compared to healthy control subjects, with a mean density of up to ∼20 million organisms per cm2 in acute lesions [22]. In addition to degrading the skin barrier, house dust mite, cockroach and scabies mite allergens with proteolytic activity were found to activate PAR2, resulting in delayed permeability barrier recovery and LB secretion following acute barrier disruption [23].
Variations in Skin Barrier Structure and Function
Although AD can affect any area of the body, it preferentially affects the flexures and the face. In babies aged less than 6 months, the face and scalp are the most common sites affected [24]. In older children, the most common sites affected are the antecubital and popliteal fossae [25,26]. Many factors could explain the areas of predisposition to AD, including the thickness of the SC and the variation in exposure to exogenous substances, such as irritants and allergens (Fig. 27.5). The eyelids and the genitals have the thinnest epidermis, followed by the flexor forearm and posterior auricular areas [27–29]. The number of cell layers in the SC also varies between different body sites and correlates with epidermal thickness [30]. A greater penetration of topical corticosteroids was observed through the skin of these areas with the thinnest epidermis [31–34].
Fig. 27.5 There is a wide difference in the thickness of the SC at different body sites. The skin sites that are not predisposed to AD have a much thicker SC where the uppermost corneocytes have been permitted to mature (left-hand panel), and as a result have a higher ‘skin barrier reserve’ (indicated in pink shading) to protect against allergen penetration. In contrast, the sites of predisposition to AD, such as the face and flexures, have the thinnest SC (right-hand panel) and can be visualized as having a very low ‘skin barrier reserve’.
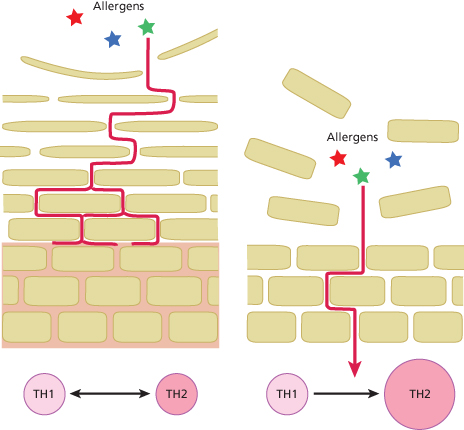
The size of the corneocytes that make up the skin barrier also varies between different body sites [35,36]. This variation correlates with skin permeability; for example, the postauricular and forehead SC were found to have the smallest corneocytes and the highest permeability compared to the upper arm, the forearm and the abdomen [36]. Furthermore, it was demonstrated that corneocytes from patients with AD are significantly smaller compared to those found in normal skin [35,37]. Taken together, variations in epidermal thickness, the layers of cells comprising the epidermal permeability barrier, and the size of the cells suggest region-specific variations in the function of the SC as a permeability barrier and therefore variations in the susceptibility to allergen penetration (see Fig. 27.5).
The thickness of the barrier and the size of the corneocytes can be attributed to the rate of desquamation. Increased desquamation results in the early loss of immature corneocytes, thereby restricting or even reducing the number of corneocyte layers, whereas reduced desquamation permits the accumulation of corneocyte layers and the extended maturation of corneocytes, which includes further flattening of their morphology, leading to a greater surface area. In agreement with this, the level of SC protease activity associated with desquamation was found to vary between different body sites [38]. This variation in activity correlated with both the thickness of the SC and the size of the corneocytes. The activity of the desquamatory proteases KLK5 and KLK7 was found to be 2–4 times higher on the cheek compared to the forearm. Notably, skin surface pH was also higher on the cheek. The surface pH of the skin is known to vary between body sites, and may provide an explanation for the differences in protease activity [39].
Measurement of TEWL is an important method of assessing barrier functionality (its ability to retain water). Nikolovski and colleagues found that the level of TEWL is associated with the thickness of the barrier, using Raman confocal microscopy [40]. Furthermore, TEWL was found to positively correlate with certain protease activities [41]. Taken together, one interpretation of these data suggests that the level of protease activity determines the rate of desquamation and thereby the structure of the barrier on a regional basis, which may involve varying degrees of PAR2 pathway activation (discussed above). Areas with low protease activity are associated with a resilient skin barrier, which retains moisture and repels allergens. On the other hand, areas with high protease activity are associated with a thin SC with low permeability barrier function – these areas can be described as having a low skin barrier reserve (see Fig. 27.5), which predisposes sites to the development of AD.
References
1 Elias P, Ahn S, Brown B et al. Origin of the epidermal calcium gradient: regulation by barrier status and role of active vs passive mechanisms. J Invest Dermatol 2002;119(6):1269–74.
2 Forslind B, Werner-Linde Y, Lindberg M et al. Elemental analysis mirrors epidermal differentiation. Acta Derm Venereol 1999;79(1):12–7.
3 Elias PM, Ahn SK, Denda M et al. Modulations in epidermal calcium regulate the expression of differentiation-specific markers. J Invest Dermatol 2002;119(5):1128–36.
4 Lee SH, Elias PM, Proksch E et al. Calcium and potassium are important regulators of barrier homeostasis in murine epidermis. J Clin Invest 1992;89(2):530–8.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


