16 Robin’s Sequence
Summary
Robin’s sequence is characterized by the triad of micrognathia, glossoptosis, and airway obstruction; in 50% of cases, a cleft palate is associated with the sequence. The cleft of the secondary palate is thought to arise from mechanical obstruction during the elevation of the lateral palatine processes from a vertical to a horizontal position. Conservative management of airway obstruction should be attempted first, with prone positioning, antireflux medications, and nasopharyngeal airway (NPA) as the mainstays of treatment. Surgical management is required in a small proportion of patients and should be preceded by airway evaluation by diagnostic endoscopy. Depending on the airway evaluation and relevant anatomy for each patient, tongue–lip adhesion, mandibular distraction, or tracheostomy may be required.
16.1 Introduction
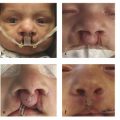
Although initially described by St. Hilaire in 1822, the French stomatologist Pierre Robin receives credit for defining this disorder in 1923. Robin wrote more than a dozen articles describing a disorder consisting of micrognathia and glossoptosis, which resulted in airway obstruction and feeding difficulties. Unlike a syndrome in which multiple findings arise from a unifying pathogenesis, Robin’s sequence arises from a chain of events, in which one anomaly leads to the next. The micro-retrognathia is thought to be due to an underlying genetic abnormality or intrauterine growth restriction related to deformation or an alteration in mandible position. This micrognathia then leads to posterior displacement of the tongue, resulting in airway obstruction and poor oral intake. Approximately half of the cases are associated with an isolated cleft palate, stemming from mechanical obstruction by the tongue during the elevation of the lateral palatine processes from a vertical to a horizontal position.
16.1.1 Epidemiology
The incidence of Robin’s sequence is reported as 1 in 8,500 to 1 in 14,000 live births, though the estimates vary between 1 in 5,000 and 1 in 50,000 births. There does not appear to be a gender discrepancy. It usually presents in isolation and is referred to as a nonsyndromic Robin’s sequence. A syndromic presentation occurs most commonly in association with Stickler’s syndrome and 22q deletion syndrome, though several other syndromes share these findings. Mortality in the early half of the 20th century was reported as high as 50%, largely due to respiratory difficulties and aspiration. With improvements in diagnosis and treatment, mortality ranges from 1.8 to 30%, and is usually related to the severity of airway distress.
16.1.2 Etiology
The cause of Robin’s sequence is unclear, and the majority of studies point to a multifactorial etiology. The micro-retrognathic mandible may be due to intrauterine growth restriction, as may occur with a multigravid pregnancy or oligohydramnios. External deformation results in constriction of mandibular growth forward. The tongue is then retropositioned in the mouth and inhibits elevation of the lateral palatine processes mechanically. Alternatively, delay of neuromuscular maturation can occur in the tongue and palate, leading to this sequence. Intrauterine exposure to ethanol and hydantoin has also been implicated.
16.1.3 Genetics
A genetic basis is heavily supported by the high incidence of twins with Robin’s sequence and a higher frequency of cleft lip and palate in family members of patients with Robin’s sequence. Deletions of 2q and 4p and duplications of 3p, 3q, 7q, 8q, 10p, 14q, 16p, and 22q are associated with cleft palate. Micrognathia is linked to deletions in 4p, 4q, 6q, and 11q and duplications of 10q and 18q. Benko has reported on an autosomal dominant locus of Robin’s sequence on chromosome 17q24.3–25.1, which codes for regulatory elements of the SOX9 gene, a key protein in chondrocyte differentiation. Rainger describes loss-of-function mutations in the SATB2 gene, which also results in micrognathia and cleft palate in humans and in a mouse model.
Sonic hedgehog and Wnt signaling drive mandibular growth from the first pharyngeal arch during embryogenesis. Absence or downregulation of β-catenin reduces mandibular growth. Inactivation of leukocyte-antigen related (LAR) family receptor protein tyrosine phosphatase (RPTP) genes mimics the features of Robin’s sequence in mouse model, resulting in aberrant jawbone and cartilage and decrease in cell proliferation in the mandible. Missense mutations in DLX5 and DLX6 in a canine model result in mandibular retrognathia and a cleft palate.
As early as 1978, Cohen recognized at least 18 syndromic diagnoses associated with Robin’s sequence. This number has grown to more than 40 syndromes. In a systematic review of the literature, Izumi found that only 40% of cases were isolated Robin’s sequence and that 60% of infants carried a syndromic diagnosis. Stickler’s syndrome is the most commonly associated, with mutations in COL2A1, COL9A, COL11A1, or COL11A2, resulting in abnormal type II or type XI collagen; these two processes colocalize in 15 to 30% of patients. About 10% of patients with Robin’s sequence are also affected by 22q deletion syndrome. This deletion in 22q11 leads to cleft palate, mandibular retropositioning, immune dysfunction, and cardiothoracic anomalies.
16.2 Diagnosis
The diagnosis of Robin’s sequence still relies on the classical description from 1923—micrognathia, glossoptosis, and airway obstruction. A cleft of the secondary palate is present in half of the cases. A multidisciplinary approach for the workup and care of these patients is required, with pediatric specialists in plastic surgery, otolaryngology, neonatology, gastroenterology, pulmonology, anesthesiology, speech pathology, and nursing all involved in diagnosis and treatment planning.
16.2.1 Presentation
Prenatal diagnosis is quite rare because micrognathia is often missed on a typical screening ultrasound. Micro-retrognathia is evident at birth and is the initial feature that prompts exploration for the diagnosis of Robin’s sequence. Mandibular hypoplasia is present in the vertical and horizontal directions, and retrogenia—posterior displacement of the chin—is also notable. The degree of glossoptosis—posterior displacement of the tongue—is due to the structure and orientation of the mandible. The tongue, while of normal size, takes up relatively more space in a volumetrically smaller oral cavity. In addition, abnormal neuromuscular control of the tongue may lead to tongue prolapse. Obstruction of the posterior pharynx by the tongue results in occlusion of the airway during inspiration. This can lead to repeated episodes of oxygen desaturation, apnea, and cyanosis. Additional energy expenditure is required during breathing to overcome these forces, with use of accessory muscles; suprasternal retractions are common during these desaturation events. Furthermore, these episodes are not continuous given they may not manifest while the infant is awake but are common during sleep, while in the supine position, or during feeding. If untreated, this may lead to hypoxia, respiratory failure, and death.
Eating is particularly challenging for these infants, given the mechanics of breathing and feeding are in direct opposition. This can result in gastroesophageal reflux and aspiration. In addition, weight gain is poor because the energy required to breathe and feed often outpaces the caloric intake that these infants can acquire. Furthermore, in the population of infants with Robin’s sequence who have a cleft palate, the inability to generate nutritive suckling in a cleft is compounded by micro-retrognathia and glossoptosis.
16.2.2 Adjunct Studies
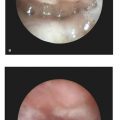
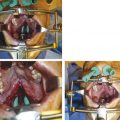
Diagnostic fiberoptic endoscopy of the airway is critical to determine optimum therapy. This is useful to determine the level of airway obstruction, which may occur at the base of tongue, epiglottis, vocal cords, or in the subglottic structures. Laryngoscopy and bronchoscopy, if possible, should be performed under anesthesia by an otolaryngologist, to evaluate for laryngomalacia and tracheal stenosis as well as to assess for vocal cord mobility and dynamic airway changes. It is critical to perform these studies in different positions, given that prone positioning may resolve airway obstruction related to the base of tongue. In 1986, Sher described four types of airway obstruction in a cohort of 33 children, as noted on diagnostic endoscopy. In type 1 and type 2 patterns, the dorsal tongue is retropositioned, whereas type 3 and type 4 described lateral pharyngeal wall motion and pharyngeal stenosis, respectively, as the cause for airway obstruction. Sher’s schema was then applied to infants with Robin’s sequence and has some predictive and therapeutic value today. Sleep studies may also be useful to evaluate for desaturation events during sleep, feeding, or position change, when the clinical diagnosis is not clear. It is not necessary to perform polysomnography routinely.
Cephalograms were used by Pruzansky to characterize mandibular shape and development in micrognathic patients; these cephalograms also give a sense of the posterior airway space. There is still controversy on whether the mandible stays persistently hypoplastic or undergoes a period of accelerated “catch-up” growth during the first few years of life. Airway obstruction often improves during this period of mandibular growth. A simple measurement of the distance between the maxilla and mandible in the horizontal occlusal plane can be used objectively to quantify the degree of mandibular hypoplasia. Both three-dimensional computed tomographic (CT) scans and three-dimensional surface photography are also useful in evaluating micrognathia and planning for surgical treatment.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


