Tumour associated
JRA, juvenile rheumatoid arthritis; PLCA, primary localized cutaneous amyloidosis.
History.
The first probable case of systemic amyloidosis was described by Fontanus (Fonteyn) in 1639, but Rokitansky is credited with identifying a lardaceous or waxy amyloid-like degradation in tissue in association with tuberculosis, syphilis and rickets more than 50 years ago [5]. During that time, Schleiden coined the term ‘amyloid’ to describe a constituent of plants [5], and Virchow used the term ‘amyloid change’ (cellulose-like) in 1854, when he observed the similar reactions of amyloid and cellulose polysaccharide to iodine and sulphuric acid [6]. In 1928, Guttman reported the first case of primary localized cutaneous amyloidosis, and Freudenthal introduced the term ‘lichen amyloidosis’ in 1930 [6].
Special stains have been used to identify amyloid since 1875 [5]. In 1930, Freudenthal implicated keratinocytes in the causation of lichen amyloidosis. Glenner et al. [7] described the AL protein in 1971, and Kumakiri and Hashimoto [4] further elucidated the role of keratinocytes in cutaneous amyloid production in 1979.
Aetiology.
Ultrastructurally, amyloid is composed of straight, non-branching fibrils of 7.5–10.0 nm in diameter, arranged haphazardly in a ‘felt-like’ array [1–3,5]. The fibrils are arranged in an antiparallel (β-pleated) configuration, which is responsible for the affinity of amyloid to cotton dyes (e.g. Congo red) [1–3,5]. Several sources of amyloid fibrillar proteins have been identified. Plasma cells secrete AL protein from intact immunoglobulin G (IgG) light chains (primarily λ) in three main disease states (immunocytic amyloidoses): primary systemic amyloidosis, amyloidosis associated with plasma cell dyscrasias (in particular, multiple myeloma) and localized nodular (tumefactive) amyloidosis of the skin or other organs [1,3,7]. Serum amyloid A (SAA) protein is a normal serum protein synthesized by the liver in association with serum high-density lipoprotein (HDL), and it behaves as an acute-phase reactant. During inflammatory states, macrophages phagocytose SAA and transform SAA into AA protein [1]. AA is present in both reactive (secondary) systemic amyloidosis and systemic heredofamilial amyloidosis [1]. Damaged and degenerating keratinocytes (e.g. due to psoralens and ultraviolet A) [8] are the source of keratinoid amyloid, which is the principal amyloid protein in PLCA [4,9]. Transthyretin, a type of prealbumin that transports thyroxine and retinol-binding protein, is the source of amyloid in some of the heredofamilial neurotropic amyloidoses [10].
Cutaneous amyloidosis in children may be due to genetic or environmental factors. Familial cutaneous lichen amyloidosis is autosomal dominant [11–15]. A form of lichen amyloidosis occurs in association with Sipple syndrome (multiple endocrine neoplasia (MEN) type 2A) [16–18], and a mutation of the RET proto-oncogene on chromosome 10 is believed to be responsible for this constellation of findings [19]. However, patients with familial cutaneous lichen amyloidosis do not have this gene mutation and therefore do not appear to be at risk of developing Sipple syndrome [20]. The genetic defect in Partington syndrome (X-linked cutaneous amyloidosis) has been localized to Xp22–p21 [21]. Macular amyloidosis is frequently an early form of lichen amyloidosis, but it may also occur due to friction from nylon brushes, backscratchers or sharp fingernails [22,23].
Pathology.
Although amyloid is derived from many different sources, the different forms of amyloid are morphologically the same on histological sections [1–3,5,6]. In haematoxylin and eosin stain, amyloid appears as amorphous, eosinophilic extracellular aggregates, without accompanying inflammation [1–3,5,6]. Special stains are helpful when minimal amounts of amyloid are present. Amyloid stains metachromatically with toluidine blue, crystal violet or methyl violet [1–3,5,6,24]. Congo red and other cotton dyes produce an apple-green birefringence in polarized light and are the most specific stains for the detection of amyloid. With thioflavine T, fluorescent microscopy reveals a yellow–green fluorescence [1–3,5,6]. Under the electron microscope, amyloid deposits are seen as straight, non-branching fibrils of 7.5–10.0 nm in diameter, arranged in a ‘felt-like’ array [1–3,5,6]. The fibril is composed of two twisted β-pleated sheet micelles, helically arranged in antiparallel conformation, as seen by X-ray crystallography [1–3,5,6].
Clinical Features
Cutaneous Amyloidosis (Box 159.1)
Primary Localized Cutaneous Amyloidosis.
Lichen amyloidosis is the most common form of PLCA. Its peak incidence is between 40 and 60 years, although in children it usually occurs in adolescence [12–14]. It is most prevalent in Chinese people residing in Taiwan, Singapore and Indonesia [12–14]. Lichen amyloidosis presents with intensely pruritic flesh-coloured, grey or yellowish-brown papules ranging in size from 1 to 10 mm. They are most commonly located on the pretibial surfaces but may also occur on the extensor surfaces of the forearms, the trunk, shoulders and sacrum (Fig. 159.1).
Box 159.1 Cutaneous Amyloidosis in Children
Primary Localized Cutaneous Amyloidosis
Major Types
- Lichenoid (sporadic/FPCA late stage)
- Macular (sporadic, ‘friction’ amyloidosis/FPCA early stage)
- Biphasic (sporadic/FPCA early stage)
Less Common Types
- FPCA associated with Sipple syndrome
- Poikiloderma-like
- Bullous
- Genodermatoses-associated macular variant
Secondary Cutaneous Amyloidosis
- Skin tumour associated
FPCA, familial primary cutaneous amyloidosis.
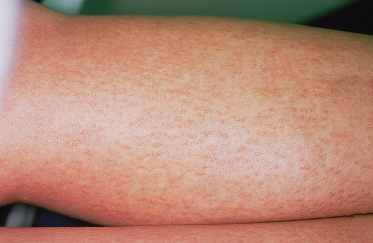
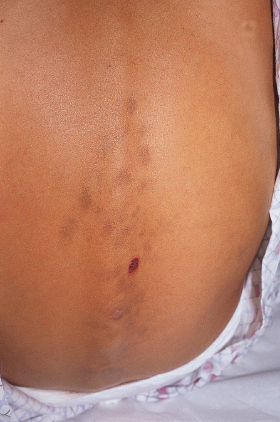
Macular amyloidosis occurs more commonly in children than lichen amyloidosis [25] and is seen more frequently in people of Latin American, Asian or Middle Eastern origin [22–25]. Typically occurring during adolescence, the macular form presents as poorly delineated hyperpigmented patches or as a linear rippling of the skin by moderately pruritic, closely aggregated greyish-brown macules. This presentation most commonly involves the lower extremities but may also involve the mid-back or arms (Fig. 159.2). Rubbing the affected area with a nylon brush or backscratcher may lead to the macular form known as friction amyloidosis [22,23].
Fig. 159.2 Macular amyloidosis. Hyperpigmented patches with superficial erosion in a 12-year-old girl.
Diffuse biphasic amyloidosis is the existence of both macular and lichen amyloidosis in the same patient. Rubbing and scratching may transform the macular to the lichenoid form, and intralesional steroids may transform the lichenoid to the macular form [3–5,22,23].
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


