History.
In 1964, Dr Robert D. Sweet [1], an English dermatologist, described eight women with ‘acute febrile neutrophilic dermatosis’. Although Dr Sweet wished to use descriptive terms to name this entity, ‘Sweet syndrome’ became its eponym [2]. In 1986, Su & Liu [3] proposed a list of two major and four minor criteria to aid in the diagnosis of Sweet syndrome. A subsequent modification by von den Driesch [4] has been widely accepted (Box 156.1).
Box 156.1 Occurrence of Sweet syndrome, according to Driesch 1994 [4]
I Classic and Idiopathic (without Prior Infection)
II Parainflammatory
Non-Infectious Disease
- Crohn disease
- Ulcerative colitis
- Rheumatoid arthritis
- Hashimoto thyroiditis
- Systemic lupus erythematosus
- Sjögren syndrome
- Mixed connective tissue disease
- Sarcoidosis
- CNS vasculitis
- Immunodeficiency syndromes
Infectious Disease
- Yersiniosis
- Typhus
- Toxoplasmosis
- Histoplasmosis
- Ureaplasmosis
- Mycobacteriosis
- Tuberculosis
- Cytomegalovirus infection
- Tonsillitis
- Hepatitis
- HIV infection
- Sepsis
III Paraneoplastic
Haematological Diseases
- Myelodysplasia
- Acute myelocytic leukaemia
- Acute myelomonocytic leukaemia
- Acute lymphoblastic leukaemia
- Chronic myelogenous leukaemia
- Multiple myeloma, hairy cell leukaemia
- Chronic lymphoblastic leukaemia
- Gammopathy
- Anaemia
- Polycythaemia
- Lymphoma (either Hodgkin or non-Hodgkin)
- Aplastic anaemia
- Fanconi anaemia
Solid Tumours
- Breast
- Stomach
- Prostate
- Uterus
- Colon and rectum
- Testis
- Lung
- Larynx
- Kidney
- Melanoma
IV Pregnancy
V Drug-Associated
Likely
- G-CSF
- ATRA
- Hydralazine
- Vaccines
Unlikely
- Antibiotics such as minocycline, nitrofurantoin and trimethoprim-sulphamethoxazole, as well as hydralazine and others
Aetiology and Pathogenesis.
The aetiology of SS is not known. The reported hypotheses concentrate on two options. One is that SS represents a classic immune complex vasculitis. Indeed, while Sweet himself considered the lack of vasculitis by means of histopathology to be characteristic, careful analysis of histological sections of classic cases of SS has revealed leucocytoclastic vasculitis in the majority of cases [5,6]. In contrast, positivity of vessels for immunoglobulin and complement factors in direct immunofluorescence of lesional skin, a hallmark of classic vasculitis, is lacking in SS [4].
The other option is that the vessel changes observed are only secondary and it is presumed that a cascade of immunological events orchestrated by T-cells leads to the enormous immigration of neutrophils into the skin, characteristic for the disease. A cascade of interleukin (IL)-1, interferon (IFN)-γ, IL-8, granulocyte colony-stimulating factor (G-CSF) and tumour necrosis factor (TNF)-α would be sufficient to explain all histopathological changes seen in SS. This would put the disease in pathogenetic proximity to pustular psoriasis and similar disorders. However, T-cell research in SS is much less advanced compared to psoriasis. Some data, however, point to a role for Th1 cells in SS [7].
Clinical Features.
Sweet syndrome has been reported worldwide and has no apparent racial predilection. It is most common among 30–60-year-old women [4]. Approximately 70 cases have been reported to involve children [8,9], almost equally distributed in both sexes from infancy through adolescence. In children below 3 years of age, male preponderance was reported in a literature analysis [9]. Familial occurrence has been reported very rarely [10].
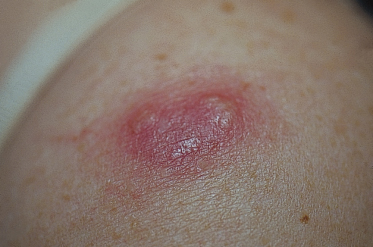
Sweet syndrome is characterized by the abrupt onset of well-demarcated, tender, erythematous papules and plaques principally located anywhere on the body. Papules and plaques can coalesce to form large plaques of up to 20 cm in diameter [4]. Severe dermal oedema may lead to the formation of vesicles or pustules [2,4] (Fig. 156.2). Scaling, crusting and eczematous changes may be present [4]. Pustules, atypical haemorrhagic bullae and ulceration (bullous pyoderma gangrenosum-like lesions, atypical Sweet syndrome) can lead to impressive clinical findings that mostly suggest local infection. This phenomenon has been described more commonly in haematological malignancies [4] and it can be difficult to diagnose SS on a clinical basis only in these cases. Ahn et al. [11] described another variant of SS with eruption of xanthoma-like lesions.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


