References
1 Slominski A, Tobin DJ, Shibahara S et al. Melanin pigmentation in mammalian skin and its hormonal regulation. Physiol Rev 2004;84:1155–228.
2 Abdel-Malek ZA, Scott MC, Furumura M et al. The melanocortin 1 receptor is the principal mediator of the effects of agouti signaling protein on mammalian melanocytes. J Cell Sci 2001;114,1019–24.
3 Rees JL. Genetics of hair and skin color. Annu Rev Genet 2003;37:67–90.
4 Lin JY, Fisher DE. Melanocyte biology and skin pigmentation. Nature 2007;445:843–50.
5 Boissy RE. Melanosome transfer to and translocation in the keratinocyte. Exp Dermatol 2003;12(suppl 2):5–12.
6 Hume AN, Ushakov DS, Tarafder AK et al. Rab27a and MyoVa are the primary Mlph interactors regulating melanosome transport in melanocytes. J Cell Sci 2007;120:3111–22.
7 Van Den Bossche K, Naeyaert JM, Lambert J. The quest for the mechanism of melanin transfer. Traffic 2006;7:769–78.
8 Seiberg M, Paine C, Sharlow E et al. Inhibition of melanosome transfer results in skin lightening. J Invest Dermatol 2000;115:162–7.
9 Betz RC, Planko L, Eigelshoven S et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet 2006;78:510–19.
10 Planko L, Bohse K, Hohfeld J et al. Identification of a keratin-associated protein with a putative role in vesicle transport. Eur J Cell Biol 2007;86:827–39.
11 Hoekstra HE. Genetics, development and evolution of adaptive pigmentation in vertebrates. Heredity 2006;97:222–34.
Disorders of Hypopigmentation
Diseases Resulting from Abnormal Melanocyte Ontogenesis
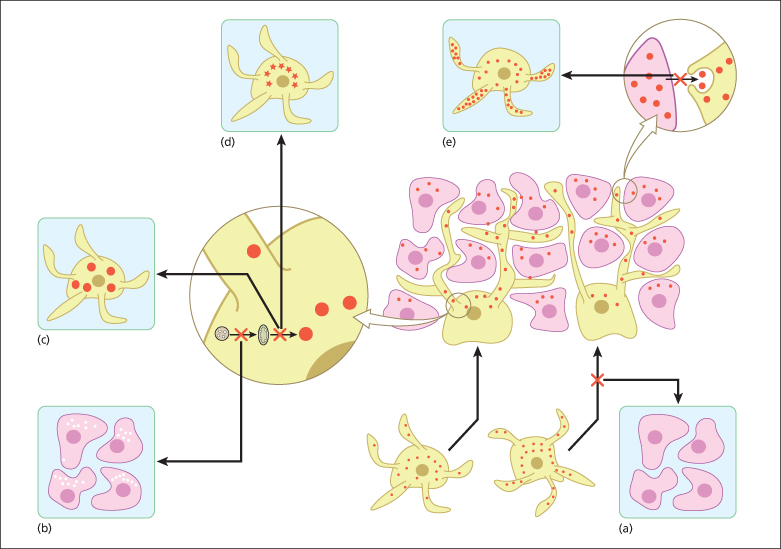
This group of pigmentation disorders mostly result from aberrant development and/or migration of melanocyte precursors to the epidermis (Fig. 138.1).
Fig. 138.1 Normal physiology of skin pigmentation and molecular defects associated with hypopigmentary disorders. During embryonic development, melanocytes (yellow) migrate to the epidermis (lower middle panel). Melanin-containing melanosomes are formed within melanocytes and are transported through dendrites to neighbouring keratinocytes (pink) where they form a cap at the upper pole of the nucleus to protect it from the deleterious effects of ultraviolet light. (a) Impaired melanocyte migration during embryogenesis results in piebaldism, characterized by patches of white skin lacking melanocytes. (b) Defects in melanin production result in lack of pigment production in oculocutaneous albinism. Chediak–Higashi syndrome (CHS), Hermansky–Pudlak syndrome (HPS) and Griscelli syndrome (GS) are characterized by dysfunction of lysosomal-related organelles. (c) The hallmarks of CHS are huge intracellular melanosomes. (d) In HPS impaired protein trafficking in melanocytes causes melanosomal dysfunction. (e) Impaired melanosome transport and capture by keratinocytes causes their accumulation in melanocytes as seen in GS.
Piebaldism
Clinical Presentation.
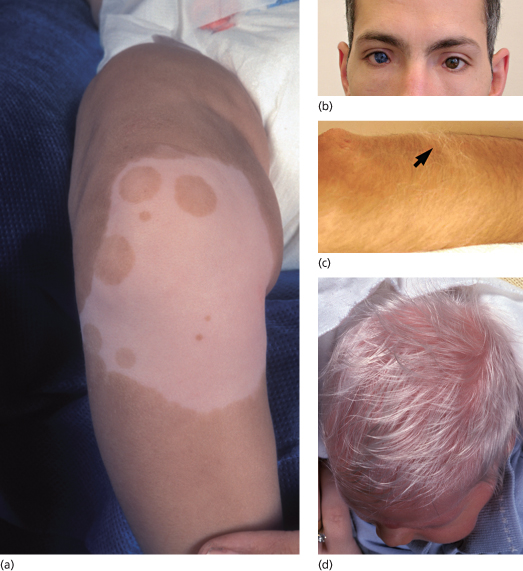
Piebaldism is a rare autosomal dominant disorder manifesting with well-demarcated irregular hypopigmentary macules [1]. The most typical and common clinical feature of the disease is a white forelock, often associated with a V-shaped area of leucoderma on the mid-forehead. The hypopigmented lesions of piebaldism have a predilection for the anterior part of the body and the midportion of the limbs (Fig. 138.2a). In contrast with vitiligo lesions, in piebaldism, lesions grow in size with the patient age and remain stable in adults. In addition, small spots of hyperpigmentation can be observed within the hypopigmentary lesions or even on the background of normal skin. Although piebaldism is usually not associated with extracutaneous manifestations, mental retardation and deafness have been reported in a number of cases [2,3].
Fig. 138.2 Disorders of hypopigmentation. (a) Well-demarcated hypopigmented macules with areas of hyperpigmentation in piebaldism. Courtesy of Dr Arie Metzker. (b) Heterochromia and dystopia canthus in Waardenburg syndrome type 1. Courtesy of Professor Peter Itin. (c) Poliosis in Waardenburg syndrome type 1 (arrow). Courtesy of Professor Peter Itin. (d) Depigmented hair and skin in oculoalbinism type 1A.
Courtesy of Dr Arie Metzker.
Pathophysiology.
The disease results from mutations in KIT, encoding c-KIT, a membranal tyrosine kinase receptor responsible for triggering cell proliferation and migration. The c-KIT receptor is very important for proper melanocyte and mast cell maturation [4]. A single nucleotide polymorphism in the KIT ligand (KITLG) gene was found to be associated with fair versus brown hair [5]. Additionally, the recent demonstration of pigmentation abnormalities resulting from the use of novel tyrosine kinase inhibitors attests to the importance of this system in the maintenance of the epidermal melanocyte population [6]. Dominant mutations in KIT result in impaired migration of melanocytes to the skin, as reflected by the absence of melanocytes and melanin in hypopigmented patches [1].
Differential Diagnosis.
Although the skin lesions in piebaldism are quite typical, the occurrence of isolated poliosis or skin hypopigmentation can suggest a number of other inflammatory (e.g. vitiligo, alopecia areata, Vogt–Koyanagi syndrome, Alezzandrini syndrome, Woolf syndrome), monogenic (e.g. tuberous sclerosis, Waardenburg syndrome) or somatic (naevus depigmentosus) disorders.
Treatment.
Epidermal cell and skin grafting have both been successfully tried in piebaldism. In contrast, phototherapy is inefficacious. Sun protection is recommended.
Waardenburg Syndrome
Clinical Features.
Waardenburg syndrome features congenital leucoderma in association with sensorineural deafness of varying severity. Areas of hypopigmentation may diminish in size or even disappear with time. Four distinct subtypes of the disease have been recognized [7]. Waardenburg syndrome type 1 is inherited in an autosomal dominant fashion and is characterized by a white forelock, alopecia, hypopigmented patches, dystopia canthorum (increased distance between the inner canthi without any change in the distance between the pupils) and heterochromia irides associated with deafness in one-third to one-half of the cases (Fig. 138.2b-c). Patients often display dysmorphic features including a broad nasal root and synophrys (medial hyperplasia of eyebrows).
Waardenburg syndrome type 2 (Klein–Waardenburg syndrome) is inherited in an autosomal dominant or recessive fashion. Clinical signs are similar to those seen in Waardenburg syndrome type 1 except for absence of facial dysmorphism and dystopia canthorum, and a higher frequency of deafness and heterochromia.
Waardenburg syndrome type 3, an autosomal dominant disorder, is much rarer than the other types and presents the same clinical manifestations as type 1 in association with musculoskeletal anomalies (e.g. syndactyly, fusion of carpal bones, etc.).
Waardenburg syndrome type 4 (Shah–Waardenburg syndrome) is inherited in an autosomal recessive fashion, features a white forelock and is accompanied by Hirschsprung disease.
Pathophysiology.
So far, mutations in six different genes have been associated with the four disease types. Type 1 and type 3 result from loss-of-function mutations in the PAX3 gene [8]. PAX3 is a transcription factor for the microphthalmia-associated transcription factor gene (MITF) which, in turn, activates a number of genes associated with melanogenesis such as TYR encoding tyrosinase [9], as well as mediating survival of melanocytes through Bcl-2. Mutations in MITF result accordingly in Waardenburg syndrome type 2. This is characterized by genetic heterogeneity (caused by mutations in different genes); for example, it was recently shown to also result from mutations in SNAI2 encoding a zinc-finger transcription factor [10]. Waardenburg syndrome type 4 is caused by mutations in at least three genes including EDNRB, encoding the endothelin-B receptor, EDN3, encoding the ligand of EDNRB, and SOX10 which, like PAX3, regulates the expression of MITF. Homozygous mutations in EDNRB and EDN3 result in Waardenburg syndrome type 4 whereas heterozygous mutations cause isolated Hirschsprung disease. Loss of SOX10 expression leads to abnormal expression of RET, which is known to cause Hirschsprung disease [9,11-13]. In addition, lack of SOX10 expression is associated with abnormal myelination and neurological signs.
Differential Diagnosis.
Tietz syndrome is characterized by skin, hair and iris hypopigmentation, hypoplasia of the eyebrows and deafness, without any photophobia or nystagmus [14].
Treatment.
Therapeutic options for the skin disease are identical to those for piebaldism. In addition, early detection of hearing loss allows for prompt intervention. Finally, surgical correction is mandatory in Hirschsprung disease.
Diseases Resulting from Abnormal Melanosome Biogenesis and Transfer
Unlike the patchy hypopigmentation typical of piebaldism and Waardenburg syndrome, mutations affecting melanosome maturation and transport (as well as melanin production, see below) are usually associated with either globally reduced or absent pigmentation of the skin, hair and eyes, with normal numbers of melanocytes in the epidermis (see Fig. 138.1).
Hermansky–Pudlak Syndrome
Clinical Features.
All eight different types of Hermansky–Pudlak syndrome are inherited in an autosomal recessive fashion [15]. All subtypes of the syndrome share common clinical manifestations including decreased pigmentation in eyes, hair and skin, easy bruisability and bleeding tendency (manifesting with menorrhagia, epistaxis, haematochezia, ecchymoses), interstitial pulmonary fibrosis and granulomatous colitis. The disorder is rare in most parts of the world except in Puerto Rico where its prevalence reaches 1:800.
Pathogenesis.
The disease results from abnormal biogenesis of lysosome-related organelles. Melanocytes are present within the skin and express a normal repertoire of genes. However, melanosome maturation is impaired and type IV melanosomes can only rarely be observed. Similarly, the platelet count is normal; however, dense bodies are absent in thrombocytes. Visceral involvement is due to partial or absent degradation of lipids resulting in accumulation of ceroid materials within the cell lysosomes. Hermansky–Pudlak syndrome is associated with mutations in eight distinct genes: HPS1 (type 1) and HPS4 (type 4) encode components of the BLOC3 lysosomal complex, which is essential for the proper formation of lysosome-related organelles; AP3B1 (type 2) encodes a subunit of the AP3 complex, which is responsible for mediating protein sorting to lysosomes; HPS3 (type 3), HSP5 (type 5) and HSP6 (type 6) encode components of BLOC2, and DTNBP1 (type 7) and BLOC153 (type 8) encode for components of BLOC1, which are all required for proper melanosome maturation [15,16].
Differential Diagnosis.
Cross–McKusick–Breen is a rare autosomal recessive disorder characterized by oculocutaneous albinism, silvery hair, growth retardation, gingival fibromatosis, spasticity, athetosis and nystagmus. Its underlying cause is unknown.
Treatment.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


