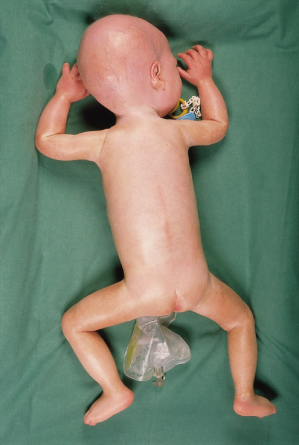
The facial manifestations include the appearance of macrocephaly with frontal bossing. The eyes appear prominent, and the nasal tip is grooved. Lips are thin and micrognathia, with a vertical midline groove in the chin, is common. Ears may protrude, and lobes are small or absent. Other findings include a pyriform chest, the development of joint stiffness, short-terminal phalanges and osteoporosis. Following fractures, bone healing may be depressed [13,16]. Radiographs demonstrate resorption of distal clavicles and phalanges. The voice is described as highly pitched. Secondary sexual characteristics fail to develop [11]. Recently described manifestations include prolonged prothrombin times, reduced joint mobility, low-frequency conductive hearing loss, and oral dysfunction [17].
Prognosis.
Most individuals succumb to cardiovascular disease, with diffuse atherosclerosis noted on autopsy [18]. Mean age at death in those with classic HGS is 12.6 years, with a range between 1.5 and 27 years [3]. Progressive intracranial vascular disease leading to strokes and seizures during the first decade of life has also been reported [19]. Intellectual functioning is at or above the normal range.
Differential Diagnosis.
Atypical HGS (including some forms of congenital HGS), mandibuloacral dysplasia, and restrictive dermopathy are most likely to be confused with classic HGS. However, in congenital HGS, progeroid manifestations are present at birth, in distinction to HGS in which they develop postnatally. Mandibuloacral dysplasia is characterized by more significant osteolysis of the clavicles, chin, and distal digits, whereas lifespan is greater than it is in HGS. Restrictive dermopathy is characterized by stiff skin with no lipodystrophy [3]. In those reported to have an autosomal recessive form of HGS, distinct manifestations include complete resorption of clavicles and distal phalanges by 5 years of age and facial abnormalities, including sparing of eyebrows, no groove on the nasal tip, full cheeks and more pronounced micrognathia. Possible examples of this condition include the children reported by Mostafa & Gabr [20], Rava [21], Franklyn [22], Maciel [23], Khalifa [24], Monu et al. [25], Hou & Wang [26], LeMerrer et al. [27], and Ramesh & Jain [28]. It is highly likely that some or all of these individuals have non-HGS mutations in LMNA or ZMPSTE24.
Treatment.
Brown [29] described the results of treating HGS patients with nutritional supplementation and growth hormones. A tripling in linear growth rate and reduction of basal metabolic rate were observed. The long-term effects of such treatment are unknown. More recently, preclinical studies in HGS cells and progeroid mouse models have shown that treatment with a farnesylation or prenylation inhibitor could reduce or prevent the formation of progerin and thus improve the outcomes in HGS patients [30,31].
References
1 Hutchinson J. Congenital absence of hair and mammary glands with atrophic condition of skin and its appendages. Med Chir Trans 1886;69:473–7.
2 Gilford H. On a condition of mixed premature and immature development. Med Chir Trans 1897;80:17–45.
3 Hennekam RCM. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A 2006;140A:2603–24.
4 Jacob KN, Garg A. Laminopathies: multisystem dystrophy syndromes. Mol Genet Metab 2006;87:289–302.
5 Eriksson M, Brown WT, Gordon LB et al. Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature 2003;423:293–8.
6 Plasilova M, Chattopadhyay C, Pal P et al. Homozygous missense mutation in the lamin A/C gene causes autosomal recessive Hutchinson–Gilford progeria syndrome. J Med Genet 2004;41:609–14.
7 Colige A, Roujeau JC, de la Rocque F et al. Abnormal gene expression in skin fibroblasts from a Hutchinson–Gilford patient. Lab Invest 1991;64:799–806.
8 Beauregard S, Gilchrest BA. Syndromes of premature ageing. Dermatol Clin 1987;5:109–21.
9 Erdem N, Gunes AT, Avci O et al. A case of Hutchinson–Gilford progeria syndrome mimicking scleroderma in early infancy. Dermatology 1994;188:318–21.
10 Yan T, Li S, Jiang X et al. Altered levels of primary antioxidant enzymes in progeria skin fibroblasts. Biochem Biophys Res Commun 1999;257:163–7.
11 Mazereeuw-Hautier J, Wilson LC, Mohammed S et al. Hutchinson–Gilford progeria syndrome: clinical findings in three patients carrying the G608G mutation in LMNA and review of the literature. Br J Dermatol 2007;156:1308–14.
12 Wang Y, Panteleyev AA, Owens DM et al. Epidermal expression of the truncated prelamin A causing Hutchinson–Gilford progeria syndrome: effects on keratinocytes, hair and skin. Hum Mol Genet 2008;17:2357–69.
13 DeBusk FL. The Hutchinson–Gilford progeria syndrome. J Pediatr 1972;80:697–724.
14 Mallory SB, Krafchik BR. Hutchinson–Gilford syndrome. Pediatr Dermatol 1990;7:317–19.
15 Gillar PJ, Kaye CI, McCourt JW. Progressive early dermatologic changes in Hutchinson–Gilford progeria syndrome. Pediatr Dermatol 1991;8:199–206.
16 Moen C. Orthopedic aspects of progeria. J Bone Joint Surg 1982;64A:542–6.
17 Merideth MA, Gordon LB, Clauss S et al. Phenotype and course of Hutchinson–Gilford progeria syndrome. N Engl J Med 2008;358:592–604.
18 Baker PB, Baba N, Boesel CP. Cardiovascular abnormalities in progeria. Arch Pathol Lab Med 1981;105:384–6.
19 Rosman NP, Anselm I. Progressive intracranial vascular disease with strokes and seizures in a boy with progeria. J Child Neurol 2001;16:212–15.
20 Mostafa AH, Gabr M. Heredity in progeria. Arch Pediatr 1954;71:163–72.
21 Rava G. Su un nucleo familiare de progeria. Minerv Med 1967;180:1502–9.
22 Franklyn PP. Progeria in siblings. Clin Radiol 1976;27:327–33.
23 Maciel AT. Brief clinical report: evidence of autosomal recessive inheritance of progeria (Hutchinson–Gilford). Am J Med Genet 1988;31:483–7.
24 Khalifa MM. Hutchinson–Gilford progeria syndrome: report of a Libyan family and evidence of autosomal recessive inheritance. Clin Genet 1989;35:125–32.
25 Monu JUV, Benka-Coker LBO, Fatunde Y. Hutchinson–Gilford progeria syndrome in siblings. Skel Radiol 1990;19:585–90.
26 Hou J-W, Wang T-R. Clinical variability in neonatal progeroid syndrome. Am J Med Genet 1995;58:195–6.
27 Le Merrer M, Guillot M, Briard M-L et al. Lethal progeroid syndrome with osteolysis. Ann Genet 1991;34:82–4.
28 Ramesh V, Jain RK. Progeria in two brothers. Aust J Dermatol 1987;28:33–5.
29 Brown WT. Progeria: a human-disease model of accelerated ageing. Am J Clin Nutr 1992;55:1222S–4S.
30 Pereira S, Bourgeois P, Navarro C et al. HGPS and related premature aging disorders: from genomic identification to the first therapeutic approaches. Mech Ageing Devel 2008;129:449–59.
31 Neilan EG. Laminopathies, other progeroid disorders, and aging: common pathogenic themes and possible treatments. Am J Med Genet 2009;149A:563–6.
Werner Syndrome
Definition.
This condition is perhaps the best example of a premature ageing syndrome. It is characterized by a combination of short stature, adult-onset sclerodermatous skin, hypogonadism, proneness to diabetes, increased incidence of malignancy and premature greying, baldness, cataracts, atherosclerosis and osteoporosis.
History.
Werner first described this condition in four siblings in a doctoral thesis in 1904 [1]. Since then over 200 patients have been reported, with recent emphasis on molecular mechanisms.
Aetiology and Pathogenesis.
The condition is clearly inherited as an autosomal recessive condition [2]. The gene maps to 8p12 [3,4]. It has been termed WRN and is a member of the RecQ family of DNA helicases [5]. The Werner protein (which is defective in Werner syndrome, WS) participates in pathways of DNA repair, recombination, transcription and replication [6–8].
Pathology.
Gawkrodger et al. [9] described skin biopsy findings in one patient. The significant findings included hyalinization of dermal collagen but no epidermal atrophy or loss of appendages. Electron microscopic evaluation demonstrated accumulation of amorphous material between normal collagen bundles.
Clinical Features.
Tannhauser [10] published 12 principal characteristics of WS. These are:
- short stature, which is unusual in that the trunk is stocky but the extremities are thin – age of onset is in the second decade
- premature greying of the hair, with age of onset in the early 20s
- premature baldness, with onset in the 20s
- scleropoikiloderma-like skin
- trophic leg ulcers
- juvenile cataracts
- hypogonadism, which is associated with reduced fertility
- tendency to develop diabetes
- calcification of blood vessels
- osteoporosis
- metastatic calcification
- tendency to occur in families.
Since that publication in 1945, it has become clear that individuals with WS are at high risk of developing cancer [11,12], with a cancer incidence over 10% [2]. The average age of diagnosis of WS is 38.7 years, with a range of 21–58 years [13]. Clinical diagnosis is based on the combination of cataracts, skin changes, short stature, and premature greying or loss of hair, with over 95% of those with homozygous mutations in WRN having these four cardinal signs [8].
Prognosis.
Average age of death is 47 years, with a range of 31–63 years. The most common causes of death are cardiovascular disease and malignancies [14]. Intellect is not impaired.
Differential Diagnosis.
Rothmund–Thomson syndrome closely resembles WS but may be distinguished by its earlier age of onset. Acrometageria and mandibuloacral dysplasia may also be confused with WS. There have also been reports of individuals with atypical WS (earlier onset of WS manifestations, generalized lipodystrophy, and skin hyperpigmentation are some of the differentiating features) who have heterozygous mutations in LMNA, the gene responsible for HGS [15].
Treatment.
Rubin et al. [16] described the results of daily treatment with human insulin-like growth factor 1, and the resultant increase in bone density in a 43-year-old woman with WS. However, no other symptoms were ameliorated.
References
1 Werner CWO. Uber Kataract in Verbindung mit Sklerodermie. Thesis. Kiel, Germany: Schmidt and Klauning, 1904.
2 Goto M, Tanimoto K, Horiuchi Y et al. Family analysis of Werner’s syndrome: a survey of 42 Japanese families with a review of the literature. Clin Genet 1981;19: 8–15.
3 Thomas W, Rubenstein M, Goto M et al. A genetic analysis of the Werner syndrome region on human chromosome 8p. Genomics 1993;16:685–90.
4 Goto M, Rubenstein M, Weber J et al. Genetic linkage of Werner’s syndrome to five markers on chromosome 8. Nature 1992;355:735–8.
5 Yu CE, Oshima J, Fu YH et al. Positional cloning of the Werner’s syndrome gene. Science 1996;272:258–62.
6 Oshima J. The Werner syndrome protein: an update. Bioessays 2000;22:894–901.
7 Bohr VA, Brosh RM Jr, von Kobbe C et al. Pathways defective in the human premature aging disease Werner syndrome. Biogerontology 2002;3:89–94.
8 Muftuoglu M, Oshima J, von Kobbe C et al. The clinical characteristics of Werner syndrome: molecular and biochemical diagnosis. Hum Genet 2008;124:369–77.
9 Gawkrodger DJ, Priestley GC, Vijayalaxmi Ross JA et al. Werner’s syndrome. Biochemical and cytogenetic studies. Arch Dermatol 1985;121:636–41.
10 Tannhauser SJ. Werner’s syndrome (progeria of the adult) and Rothmund’s syndrome: two types of closely related heredofamilial atrophic dermatoses with juvenile cataracts and endocrine features: a critical study of five new cases. Ann Intern Med 1945;23:559–626.
11 Rosen RS, Cimini R, Coblenz D. Werner’s syndrome. Br J Radiol 1970;43:193–8.
12 Bjornberg A. Werner’s syndrome and malignancy. Acta Dermatol Venereol (Stockh) 1976;56:149–50.
13 Epstein CJ, Martin MG, Schultz AL et al. Werner’s syndrome: a review of its symptomology, natural history, pathologic features, genetics, and relationship to the natural ageing process. Medicine 1966;45:177–221.
14 Thweatt R, Goldstein S. Werner syndrome and biological ageing: a molecular genetic hypothesis. Bioessays 1993;15:421–6.
15 Doh YJ, Kim HK, Jung ED et al. Novel LMNA gene mutation in a patient with atypical Werner’s syndrome. Korean J Intern Med 2009;24:68–72.
16 Rubin CD, Reed B, Sakhaee K et al. Treating a patient with the Werner syndrome and osteoporosis using recombinant human insulin-like growth factor. Ann Intern Med 1994;121:665–8.
Cockayne Syndrome (see Chapter 135)
Cockayne syndrome is a heterogeneous condition. The classic form is characterized by postnatal growth deficiency, cataracts, pigmentary retinopathy, skin photosensitivity, hearing loss and mental retardation. A severe form has been described, and includes prenatal growth deficiency and earlier onset of symptoms. A mild form has also been described, and includes normal growth and intelligence. In addition, Forsythe et al. [1] recently described four siblings with a Cockayne-like syndrome who also had thrombocytopenia and nephrosis, but no increase in photosensitivity. The authors suggested that this was probably a unique DNA repair disorder.
Reference
1 Forsythe E, Wild R, Sellick G et al. A novel DNA repair disorder with thrombocytopenia, nephrosis, and features overlapping Cockayne syndrome. Am J Med Genet 2009;149A:2075–9.
Rothmund–Thomson Syndrome (see Chapter 136)
Rothmund–Thomson syndrome is an inherited disorder whose phenotype includes early childhood-onset poikiloderma, with occasional occurrence of cataracts and skeletal anomalies. An increased risk of malignancy is one of the manifestations.
Dyskeratosis Congenita (see Chapter 136)
Dyskeratosis congenita is a heterogeneous condition characterized by skin manifestations, premature greying, tooth loss, osteoporosis, and increased frequency of malignancy.
Conditions with Skin Atrophy/Lipoatrophy
Wiedemann–Rautenstrauch Syndrome
Syn.
Neonatal progeroid syndrome
Definition.
Wiedemann–Rautenstrauch syndrome (WRS) is a premature ageing syndrome characterized by intrauterine growth retardation, short stature, typical facial appearance, natal teeth, lipoatrophy and paradoxical caudal fat accumulation. Over 30 patients have been described.
History.
Rautenstrauch & Snigula [1] first described two sisters with a progeria-like syndrome in 1977. Wiedemann [2] described two additional patients 2 years later; Devos et al. [3] suggested the acronym WRS.
Aetiology.
This is almost certainly an autosomal recessive trait in that both affected siblings and parental consanguinity have been described [1,3–5]. The basic genetic defect is unknown, although Beavan et al. [6] described deficient decorin expression in one patient originally described by Rautenstrauch & Snigula. Decorin is a small proteoglycan that may interact with collagen I and II to influence the rate of fibril formation. However, the authors did not consider a decorin deficiency to be the primary defect because decorin expression returned to normal levels in adolescence in that patient. Mazzarello et al. [7] described reduced thymidine kinase activity in skin fibroblasts, suggesting a DNA metabolism defect. Mutations in LMNA, ZMPSTE24 and ERCC8 have been sought, but not found [8,9]. Therefore the cause of WRS remains elusive.
Pathology.
Brain examination by Martin et al. [10] on the patient described by Devos et al. [3] showed a sudanophilic leucodystrophy with tigroid streaks. Hagadorn et al. [11] did not find this in their patient, and suggested that heterogeneity may exist. Ulrich et al. [12] described absence of mature myelin in their patient’s brain. They also suggested that WRS may be heterogeneous, neuropathologically and probably genetically. Skin biopsy done on one patient demonstrated only marked hypoplasia of corium [13]. Proliferation rate of fibroblasts was one-half of that of normal control patients.
Clinical Features.
Affected individuals have intrauterine growth retardation with failure to thrive and short stature. The progeroid appearance is apparent at birth, with the phenotype consisting of a pseudo-hydrocephaloid appearance (although the occipitofrontal circumference is within normal limits), sparse hair, prominent scalp veins, widened anterior fontanelles and malar hypoplasia. One to four natal teeth are almost always present, with these teeth lost and subsequent dentition delayed. Skin is dry, thin and wrinkled. Hands and feet appear large. Generalized lipoatrophy is present, although paradoxical caudal fat accumulation occurs during childhood. One child also had fat accumulation in the axillae and on the proximal portion of the digits [13]. Feeding difficulties are common. Over time, the nose appears beaked. Cognitive impairment is usually present, with mild to severe degrees of impairment reported. Joint contractures, cardiac defects, hydronephrosis and congenital hearing loss are occasional manifestations. Some patients have been reported to have elevated insulin and triglyceride levels [14], but this is not a consistent manifestation [15].
Prognosis.
Longevity is unknown, with the oldest patient being 17 years old at the time of the report [16]. The mean age of survival, however, is 7 months [16]. As noted above, cognitive impairment may be present, ranging from mild to severe.
Differential Diagnosis.
Natal teeth are present in Hallerman–Streiff, Ellis–van Creveld and Ullrich Fremerez–Dohna syndromes. Progeroid facial appearance at birth can occur in Hallermann–Streiff, Berardinelli–Seip, Bamatter and DeBarsy syndromes. Paradoxical caudal fat accumulation occurs in congenital disorders of glycosylation. One child thought to have WRS was subsequently found to have somatic mosaicism for triploidy and tetraploidy, so a skin biopsy for karyotyping should be considered, particularly when unusual manifestations such as cardiac defects and syndactyly of the fingers are present [17]. The syndrome described by Petty et al. [18] should be distinguished from WRS.
References
1 Rautenstrauch T, Snigula F. Progeria: a cell culture study and clinical report of familial inheritance. Eur J Pediatr 1977;124:101–11.
2 Wiedemann HR. An unidentified neonatal progeroid syndrome: follow up report. Eur J Pediatr 1979;130:65–70.
3 Devos EA, Leroy JG, Fryns JP et al. The Wiedemann–Rautenstrauch or neonatal progeroid syndrome: report of a patient with consanguineous parents. Eur J Pediatr 1981;136:245–8.
4 Castincyra G, Panal M, Presas HL et al. Two sibs with Wiedemann–Rautenstrauch syndrome: possibilities of prenatal diagnosis by ultrasound. J Med Genet 1992;29:434–6.
5 Pivnick EK, Angle B, Kaufman RA et al. Neonatal progeroid (Wiedemann–Rautenstrauch) syndrome: report of five new cases and review. Am J Med Genet 2000;90:131–40.
6 Beavan LA, Quentin-Hoffmann E, Schonherr E et al. Deficient expression of decorin in infantile progeroid patients. J Biol Chem 1993;268:9856–62.
7 Mazzarello P, Verri A, Mondello C et al. Enzymes of DNA metabolism in a patient with Wiedemann–Rautenstrauch progeroid syndrome. Ann NY Acad Sci 1992;663:440–1.
8 Cao H, Hegele RA. LMNA is mutated in Hutchinson–Gilford progeria (MIM 176670) but not in Wiedemann–Rautenstrauch progeroid syndrome (MIM 264090). J Hum Genet 2003;48:271–4.
9 Hou J-W. Natural course of neonatal progeroid syndrome. Pediatr Neonatol 2009;50:102–9.
10 Martin JJ, Ceuterick CM, Leroy JG et al. The Wiedemann–Rautenstrauch or neonatal progeroid syndrome. Neuropathological study of a case. Neuropediatrics 1984;15:43–8.
11 Hagadorn JI, Wilson WG, Hogge WA et al. Neonatal progeroid syndrome: more than one disease? Am J Med Genet 1990;35:91–4.
12 Ulrich J, Rudin C, Bubl R et al. The neonatal progeroid syndrome (Wiedemann–Rautenstrauch) and its relationship to Pelizaeus–Merzbacher’s disease. Neuropath Appl Neurobiol 1995;21:116–20.
13 Rudin C, Thommen L, Fliegel C et al. The neonatal pseudohydrocephalic progeroid syndrome (Wiedemann–Rautenstrauch). Eur J Pediatr 1988;147:433–8.
14 Dinleyici EC, Tekin N, Dinleyici M et al. Clinical and laboratory findings of two newborns with Wiedemann–Rautenstrauch syndrome: additional features, evaluation of bone turnover and review of the literature. J Pediatr Endocrinol Metab 2008;21:591–6.
15 O’Neill B, Simha V, Kotha V et al. Body fat distribution and metabolic variables in patients with neonatal progeroid syndrome. Am J Med Genet 2007;143A:1421–30.
16 Arboleda H, Arboleda G. Follow-up study of Wiedemann–Rautenstrauch syndrome: long-term survival and comparison with Rautenstrauch’s patient “G”. Birth Defects Res A 2005;73:562–8.
17 Karteszi J, Kosztolanhi G, Czako M et al. Transient progeroid phenotype and lipodystrophy in mosaic polypoidy. Clin Dysmorph 2006;15:29–31.
18 Petty EM, Laxova R, Wiedemann HR. Previously unrecognized congenital progeroid disorder. Am J Med Genet 1990;35:383–7.
DeBarsy Syndrome
Definition.
DeBarsy syndrome phenotype consists of progeroid appearance, growth and mental retardation, cutis laxa, corneal clouding and athetoid movements.
History.
DeBarsy et al. [1] described the combination of prenatal growth retardation, skin laxity, minor craniofacial anomalies, cloudy corneae, large anterior fontanelle with delayed closure, and athetoid movements in a single girl in 1968. Since then, over 25 patients have been described, including three as unknown cases [2–12]. Some of the cases reported as having DeBarsy syndrome, however, probably have a distinct condition [3,8].
Aetiology and Pathogenesis.
This is clearly inherited as an autosomal recessive trait based on several examples of affected siblings. The basic genetic defect is unknown, although Karnes et al. [11] found that fibroblasts contained reduced levels of elastin mRNA, which may in turn be due to decreased transcription or increased degradation of mRNA.
Pathology.
Skin biopsy performed by DeBarsy et al. [1] demonstrated normal epidermis, but thinner than normal dermis. The collagen fibres were described as having few fasciculations and elastic fibres were thin, short and decreased in number. Karnes et al. [11] described decreased elastic fibres, as well as skin biopsy changes over time. In the neonatal period, their patient had hyperkeratosis and papillomatosis of the epidermis, with a deficiency of elastic fibres. At 10 months, the epidermis was normal and the dermis thin. The adnexal structures were located at the dermoepidermal junction instead of their usual location. Elastic fibres remained decreased in number and size.
Electron microscopy demonstrated variability in the collagen bundle size, as well as increased microfibrillar component of elastin and thinning of the amorphous component of elastin.
Clinical Features.
Children with DeBarsy syndrome generally have intrauterine growth retardation and subsequent slow growth. Corneal clouding or cataracts are virtually constant and the facial phenotype is described as progeroid. Specifically, these children have a prominent forehead, small nose with upturned nares, and thin lips. The eyes usually appear deeply set. The ears are described as large and dysplastic, with a relatively unfolded helix. Hypotonia is a virtually constant finding, as is mild cutis laxa and thin wrinkled skin, particularly of the extremities. Small joints are often hyperflexible, and hip dislocation or club-foot occur frequently. Athetoid movements are common, but it is unclear whether these are present in all affected individuals. Cognitive impairment is present, being severe in almost half [12]. No child was reported to have normal development.
Prognosis.
Lifespan is unknown, because most cases were reported when the child was an infant. The oldest reported individual was 24 years old at the time of the report and did not have any life-threatening health problems [3]. However, it has been questioned whether this individual and his siblings have DeBarsy syndrome, particularly because they have a different facial phenotype, had normal birthweights, and had hyper- and hypopigmented skin patches, which are not seen in other individuals with DeBarsy syndrome [12].
Differential Diagnosis.
Apparently some children with DeBarsy syndrome are initially diagnosed as having WRS, but the presence of natal teeth and caudal fat accumulation in WRS should distinguish between the two. The other premature ageing syndromes should also be considered in the differential diagnosis.
Treatment.
No treatment is known.
References
1 DeBarsy AM, Moens E, Dierckx L. Dwarfisms, oligophrenia and degeneration of the elastic tissue in skin and cornea: a new syndrome? Helv Pediatr Acta 1968;23:305–13.
2 Schierenberg M, Donne W, Schiafone P et al. De Barsy–Moens–Dierckx-Syndrom: ein ungewohnlicher Verlauf bei einem Fruhgeborenen. Klin Padiatr 1994;206:444–6.
3 Kunze J, Majewski F, Montgomery P et al. DeBarsy syndrome: an autosomal recessive, progeroid syndrome. Eur J Pediatr 1985;144:348–54.
4 Stanton RP, Rao N, Scott CI. Orthopaedic manifestations in De Barsy syndrome. J Pediatr Orthop 1994;14:60–2.
5 Harrod MJ, Keele D, Stevenson RE. The De Barsy syndrome. Proc Greenwood Genet Cent 1984;3:134.
6 Hoefnagel D, Pomeroy J, Wurster D et al. Congenital athetosis, mental deficiency, dwarfism, and laxity of skin and ligaments. Helv Pediatr Acta 1971;26:397–402.
7 Burck U. De Barsy-Syndrom: eine weitere Beobachtung. Klin Padiatr 1974;186:441–4.
8 Riebel T. DeBarsy–Moens–Dierckx-Syndrom: Beobachtung bei Geschwistern. Mschr Kinderheilk 1976;124:96–8.
9 Siedel H, Stengel-Rutkowski S, Schimanek P et al. Non-chromosomal dysmorphic syndromes (MCA/MR syndromes). 1. Similar abnormal phenotype in two mentally retarded brothers. Dysmorph Clin Genet 1987;1:101–8.
10 Saul R (ed). Unknown case (R.F.W.). Proc Greenwood Genet Cent 1983;2:70–1.
11 Karnes PS, Shamban AT, Olsen DR et al. De Barsy syndrome: report of a case, literature review, and elastic gene expression studies of the skin. Am J Med Genet 1992;42:29–34.
12 Kivuva EC, Parker MJ, Cohen MC et al. De Barsy syndrome: a review of the phenotype. Clin Dysmorphol 2008;17:99–207.
Acrometageria
Definition.
This is a presumed spectrum of phenotypes that encompasses acrogeria, which primarily affects the hands and feet, and metageria, which involves the limbs as well as other structures. This entity is clearly heterogeneous.
History.
Gottron [1] first described a progeroid syndrome that primarily affected the skin of the hands and feet. Since then, over 50 cases have been described. In 1974, Gilkes et al. [2] described two patients with phenotypes similar to but believed to be distinct from acrogeria and HGS or WS. In 1992 Greally et al. [3] described a boy with features of both acrogeria and metageria, and hypothesized that acrogeria and metageria are part of the phenotypic spectrum of a single disease entity. They suggested acrometageria as the name for this condition. It is now clear that this is a group of similar, but genetically distinct, conditions.
Aetiology and Pathogenesis.
Although most affected individuals are the only family members affected, Kaufman et al. [4] described a pedigree consistent with autosomal dominant inheritance, in which one individual clinically had metageria and two others had acrogeria. The other cases therefore could represent fresh mutations. Although a defect in collagen III synthesis was suggested by Pope et al. [5] and Bouillie et al. [6], Bruckner-Tuderman et al. [7] and Blaszczyk et al. [8] did not find abnormal collagen III levels. It is likely that acrometageria is heterogeneous and that some patients with collagen III deficiency (which also causes Ehlers–Danlos IV) have an acrogeroid phenotype [9,10]. Hunzelmann et al. [11] described deficiency of type 1 collagen in a man with metageria and his sister with acrogeria.
Pathology.
Meurer et al. [12] examined the skin in a man diagnosed with acrogeria. Subcutaneous fat was diminished, dermal papillae were flattened and there was orthokeratotic hyperkeratosis. Collagen fibre number was decreased, whereas elastin fibres were increased, although they were fragmented in appearance. The granular endoplasmic reticulum was dilated, so cells appeared vacuolized. In the vacuoles, as well as extracellular areas, pseudo-elastin was present. Bruckner-Tuderman et al. [7] also examined the skin from a patient with acrogeria, but noted differences between biopsy sites. These differences included reduced thickness of the dermis and more collagen bundle abnormalities in the foot specimen compared with the axilla specimen. A third report of skin biopsy results did not note any abnormalities in a specimen taken from the buttock [3]. Tajima et al. [13] described late-onset focal dermal elastosis in a patient.
Clinical Features.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


