The variety of cytogenetic abnormalities seen in PM explains the heterogeneity of systemic features associated with PM. The common feature of pigmentary disturbance may represent a non-specific result of co-migration of genetically different cell lines (as in chimeras). Alternatively it is possible that the many different chromosomal anomalies reported in PM disrupt expression or function of a multiplicity of pigmentary genes scattered across the genome [15]. Pigmentary genes control various processes, such as melanoblast migration from the neural crest in fetal life and melanocyte function, including synthesis, transport and degradation of melanosomes. Furthermore, melanocyte function can be disrupted by genetic abnormalities affecting the surrounding milieu, which includes keratinocytes, intercellular matrix, growth factors, etc. Animal models (particularly mouse and Drosophila) have identified numerous pigmentary genes, many of which have human homologues implicated in pigmentary disorders. Recent reviews are available for both mice [19] and humans [20].
Pigmentary differences in PM might arise by disruption of any of these genes. For example, this idea is supported by the finding of selective suppression of eumelanin synthesis in hypopigmented compared to normal-looking skin in one HI patient, although the specific gene disrupted in this case was undetermined [21]. Pellegrino et al. [22] implicated the P gene in PM cases with cytogenetic abnormalities involving 15q, and Kosaki et al. [23] recently demonstrated somatic mosaicism of p63 in a case of PM with associated split hand-foot malformation.
A number of additional genetic mechanisms have been proposed which may modulate the somatic expression of genes during development, potentially giving rise to mosaic patterns of pigmentation. These include the following.
- Transposons. Transposable elements of retroviral origin (transposons) occur in the normal genome. Some can regulate gene activation and silencing, and hence modulate the activity of adjacent genes by processes involving methylation or demethylation. Based on animal models, Happle [24] hypothesized that partial activation or silencing of pigmentary genes by transposons present in some cells could give rise to PM. However, this is currently unsubstantiated in humans.
- Genetic imprinting. Imprinting, in which phenotype is determined by the parental origin of the autosomal gene, may be relevant to PM. For example, Hatchwell et al. [16] observed that in cases of HI with balanced X-autosome translocations, the translocated chromosomal material is most commonly of paternal origin.
- Phenotypic reversion. Mice heterozygous for certain dominant mutations at the Kit locus display an almost completely white coat. However, in one study, 3.6% of such mice exhibited patches of wild-type pigmentation, termed phenotypic reversion [25]. Melanocyte cell lines derived from these patches showed somatic reversion from the mutant to the wild-type Kit allele. The relevance of phenotypic reversion to patients with PM is unconfirmed.
Histopathology.
Histology is of limited value in diagnosis. In patients with LWNH, diffuse basal layer hyperpigmentation is seen [3,26]. Lichenoid changes and pigmentary incontinence with melanophages are not generally seen in either HI or LWNH, as is to be expected since there is no preceding inflammation, in contrast to IP [2].
In keeping with cytogenetic heterogeneity and the idea of pigmentary gene disruption, a variety of changes have been reported with DOPA staining or ultrastructural examination [27–30]. In HI, decreased numbers of melanocytes as well as defective pigment production are described. Melanocytes may appear more rounded with reduced or absent dendrites. Premelanosomes may be reduced in number and small in size. Increased number of nerve endings in contact with melanocytes has also been seen, the significance of which is uncertain [31].
Clinical Features.
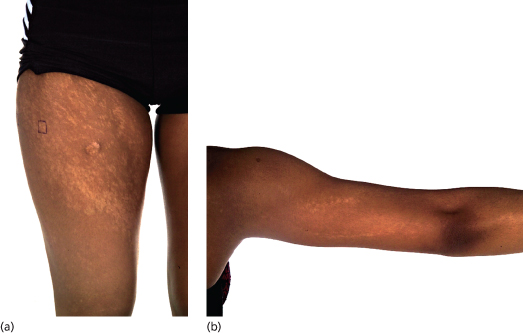
Patients with PM present with mosaic patterns of cutaneous dyspigmentation, classically with hypo- or depigmented skin in HI (Fig. 131.2) and hyperpigmentation in LWNH (Fig. 131.3). Rarely patients show both hypo- and hyperpigmentation, further supporting the use of the term PM. The pigmentary pattern is most commonly along the lines of Blaschko, although other mosaic patterns may be observed including phylloid (leaf-like), chequerboard and patchy pigmentation without midline separation [10]. In some cases dyspigmentation can be subtle and in patients referred with systemic abnormalities such as neurodevelopmental delay and epilepsy, it is important to examine the entire skin. A Wood’s light ultraviolet source may facilitate identification of hypopigmentation analogous to detection of hypopigmented ash-leaf macules in tuberous sclerosis.
Fig. 131.2 (a,b) Eighteen-year-old girl with linear and streaky hypopigmentation (rectangle marked on lateral thigh corresponds to biopsy site from which trisomy 20 mosaicism was subsequently identified in cultured keratinocytes).
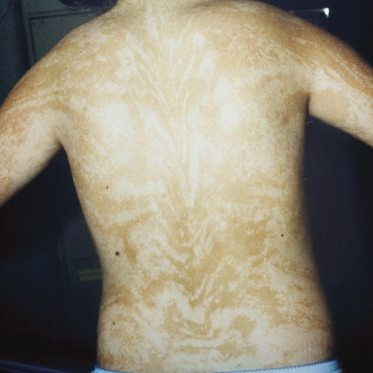
A variety of extracutaneous features has been reported, in keeping with the genetic heterogeneity of PM [9,17,32–45] (Table 131.1). Most frequent are neurological deficit including learning difficulties, autism [37] and epilepsy, and musculoskeletal defects, although almost any organ system may be involved. Most authorities would only diagnose naevoid dyspigmentation as PM in the presence of extracutaneous anomalies, ideally with confirmation of cytogenetic mosaicism. The frequency of different presentations is difficult to ascertain because of publication bias. Patients with cutaneous pigmentary patterns suggestive of mosaicism are likely to go unreported if there are no clinically apparent systemic abnormalities, or alternatively if the associated features are already well described in the literature.
Table 131.1 Extracutaneous features associated with pigmentary mosaicism
| Organ system | Associated features |
| Neurological | Learning difficulties, epilepsy, dyskinesias including choreo-athetosis, hypotonia, spasticity, autism, microcephaly, macrocephaly, agenesis of corpus callosum, cerebral atrophy |
| Musculoskeletal | Facial asymmetry/dysmorphism, cleft lip/palate, short stature, limb asymmetry, limb defects, pectus excavatum/carinatum, scoliosis, syndactyly, clinodactyly, brachydactyly, talipes equinovarus, pes planus, congenital hip dislocation, breast asymmetry, torticollis |
| Hearing/ears | Deafness, pre-auricular pit, asymmetry |
| Ophthalmological | Ptosis, nystagmus, symblepharon, dacrostenosis, strabismus, nystagmus, corneal opacities, cataract, myopia, amblyopia, microphthalmia, buphalmos, heterochromia, scleral melanosis, retinal dyspigmentation, retinal degeneration, cortical dysplasia |
| Skin | Café-au-lait macules, aplasia cutis, impaired sweating, accessory nipples |
| Nails | Ridging, hypoplasia, anonychia |
| Hair | Sparse hair, hypertrichosis, colour variegation |
| Teeth | Enamel defects, hamartomatous dental cusps, hypodontia, caries |
| Cardiac | Tetralogy of Fallot, VSD, PDA, AS, PS, conduction defects |