Aetiology.
Several risk factors exist for porokeratosis including genetic inheritance, ultraviolet radiation (including electron beam, radiation therapy and artificial ultraviolet radiation) and immunosuppression. Porokeratosis has been observed in patients with HIV and lymphomas as well as patients on immunosuppressive medications for transplant or autoimmune diseases. Most patients who develop porokeratosis have less pigmented skin, although porokeratosis can be seen in more darkly pigmented individuals. The formation of non-melanoma skin cancers including squamous cell carcinomas and basal cell carcinomas have been reported for all types of porokeratosis although linear porokeratosis and large lesions of long duration appear to have the greatest risk. Reports of malignant transformation into skin tumours range from 7.5% to 11.6%. Instability in chromosome 3 has been associated with development of malignancy in cultured fibroblasts derived from porokeratosis lesions.
Three genetic loci have been identified to date in patients with DSAP: 2q23.2-24.1 (DSAP1), 15q25.1-26.1 (DSAP2) and 1p31.3-p31.1 (DSAP3) and 18p11.3. Two candidate genes at the DSAP1 locus [SSH1 (slingshot homologue-1) and SART3 (squamous cell carcinoma antigen recognized by T cells 3)] were characterized. It remains to be determined which one of the two genes or if a different gene is the DSAP-causing gene at this locus. Similarly, on DSAP3 eight candidate genes were sequenced, but found to be negative for functional sequence variants. Two genetic loci for other two subtypes of porokeratosis were also mapped, one for disseminated superficial porokeratosis (DSP) on 18p11.3 and one for PPPD on 12q24.1–24.2.
In 2010, microarray analysis using an Illumina platform from porokeratosis patients’ lesional and non-lesional skin identified three candidate genes: SART3, SSH1 and ARPC3 (actin related protein 2/3 complex, subunit 3) which were upregulated in lesional skin. Keratin 6a was identified as a specific biomarker for porokeratotic keratinocytes as it was the most significantly upregulated gene in the nine patient samples. Previous work by Hivnor using an Affymetrix platform identified 10 upregulated genes including keratins 6A, 6B, 16, 17, S-100A7 (S-100 calcium binding proteinA7/psoriasin), A8, A9, FABP5 (fatty acid binding protein 5, psoriasis-associated), GJB2 (gap junction protein beta 2/connexin 26) and SPRP1A (small proline-rich protein 1A).
The subtypes of porokeratosis arise at different time points in an individual’s lifetime with linear porokeratosis and PPPD occurring at any time between birth and adulthood, while PM develops in childhood and DSAP typically occurs in third or fourth decade of life.
Clinical Variants
Porokeratosis of Mibelli
This entity was first reported in 1889 by Vittorio Mibelli in a 21-year-old patient with an affected father and sibling, as multiple annular and gyrate plaques with central atrophy and elevated keratotic borders containing a longitudinal furrow. Mibelli coined the term ‘porokeratosis’ to emphasize what he believed to be representative features of the lesion: abnormal keratinization and origination within the pores of sweat ducts. Porokeratosis of Mibelli lesions are typically asymptomatic or very slightly itchy, light brown keratotic papules that develop in childhood. While there is usually an autosomal dominant inheritance pattern, this entity can also be acquired. As the lesion progresses, papules slowly expand to form an annular plaque with a raised border and atrophic centre. These lesions may expand rapidly if the patient becomes immunosuppressed. There are reports of patients developing this entity after diagnosis of HIV. Frequently there is a history of antecedent burn, radiation therapy or other trauma to the area where the lesion first appears. For PM, the main differential diagnoses include guttate psoriasis and warts. There are rare reports of cutaneous T-cell lymphoma mimicking porokeratosis so this should be considered. Use of dermoscopy to identify the hyperkeratotic border has been proposed recently. Under dermoscopy a white peripheral rim, which corresponds to cornoid lamellae, is the essential pathognomonic feature for diagnosis.
Disseminated Superficial Actinic Porokertosis
Four years after Mibelli described porokeratosis, Respighi described the disseminated superficial variant. In 1966, Chernosky defined DSAP (also known as porokeratosis of Chernosky) as a distinct clinical entity characterized by small inconspicuous lesions occurring on sun-exposed areas in adults. A subsequent detailed light microscopic analysis of 35 clinically varied cases was published by Reed and Leone in 1970. They observed that the majority of lesions were not associated with ostia of eccrine or pilosebaceous ducts, and asserted that the well-accepted term ‘porokeratosis’ was a misnomer. A more precise term was not proposed. This entity is composed of multiple small scaly brown keratotic papules with raised borders which occur on the extensor surfaces. Lesion number ranges from a few to several hundred but typically is greater than 50 lesions. They may be asymptomatic or slightly itchy. Half of patients note exacerbation of their lesions during the summer months. Facial lesions are uncommon and occur in less than 15% of patients. Most patients who develop DSAP are women in their fourth and fifth decades with an extensive history of ultraviolet radiation exposure (such repeated tanning/tanning bed exposure or ultraviolet exposure from phototherapy). Immunosuppression also predisposes to DSAP. For DSAP the main differential diagnoses include psoriasis, stucco keratoses, actinic keratoses, squamous cell carcinoma, warts and Darier disease.
In 2002, two loci for DSAP were mapped to 12q23.2–24.1 and 15q25.1–26.1 in two Chinese families and in 2004 an additional locus was mapped to chromosome 18p11.3. However, no disease genes for DSAP have been identified so far.
In 2009, a new variant was described with neutrophilic pustules within the cornoid lamellae which corresponds to pustules on the outer rim clinically. This was the second report of porokeratosis with pustules.
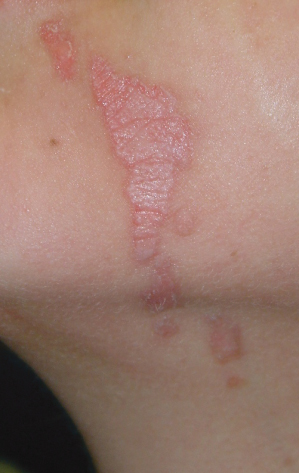
Linear Porokeratosis
This rarer subtype was historically described in 1974 by Rahbari as an entity distinct from PM. These lesions typically occur in infancy or childhood and do not appear to be inherited. They are red–brown linear keratotic papules and annular plaques often in a Blaschkoid distribution (Fig. 126.2). Nail dystrophy has been associated with this disorder. Loss of heterozygosity may account for higher risk of malignant degeneration within these lesions. These lesions may also have increased risk for p53 mutations. For linear porokeratosis, the main differential diagnoses include linear verrucous epidermal naevus, lichen striatus, incontinentia pigmenti, linear lichen planus, linear psoriasis, linear Darier disease and warts.
Porokeratosis Palmaris and Plantaris Disseminata
This is a variant originally described by Guss in 1971. These are small keratotic papules, which are sometimes itchy, on the palms and soles which occur during adolescence and early adulthood. These may become generalized and involve the trunk and extremities. The appearance is similar to DSAP except that the lesions are not limited to sun-exposed areas. Mucosal lesions have been noted occasionally. Squamous cell carcinomas are reported to develop within these lesions. These lesions can be transmitted in an autosomal dominant mode or caused by immunosuppression. Sudden develop of these lesions should prompt a search for internal malignancy. Differential diagnoses include palmo-plantar keratodermas, calluses and warts.
In 2003, a locus was located at chromocome 12q24.1–24.2 but no disease genes or mechanisms were identified. Two candidate genes, SSH1 and SART3, with uncertain signifiance were isolated from one screen. Flow cytometry of cells from lesional skin have shown abnormal DNA ploidy.
Punctate Porokeratosis
Punctate porokeratosis is manifested by multiple small (0.2–1.0 cm) firm flesh-coloured hyperkeratotic papules on the palms and soles of adults. Papules are firmly attached at their bases. There is no inheritance pattern (although both sporadic and autosomal dominant forms have been reported) and usually these are associated with other forms of porokeratosis. Clinically, these lesions resemble punctate porokeratotic keratoderma which is considered an sign of internal malignancy. Differential diagnoses include punctate, palmo-plantar keratoderma (Buschke–Fischer disease), acrokeratoelastoidosis, punctate keratosis of palmar creases, focal acral hyperkeratosis, calluses and warts.
The aetiology of this disease is not certain. In one study, keratins 6 and 16 were present in lesional skin. Lesions do not resolve spontaneously.
Porokeratosis Ptchyotropica
This lesser known variant is characterized by circumferential perianal plaques. These lesions have the typical cornoid lamella histology but with underlying amyloid deposition. Differential diagnoses include inverse psoriasis, chronic contact or irritant dermatitis, acrodermatitis enteropathica, necroytic migratory erythema, chronic intertrigo, Darier disease and Hailey–Hailey disease.
Genital Porokeratosis
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


